Introduction
- In our project we needed to find or create a transmembrane fusion protein capable of linking the Claudine-4, a biomarker found on the precancerous cells inducting the pancreatic ductal adenocarcinoma (PDAC).
- After several research on scientific literature and on the iGEM previous Wiki, we found numerous fusion proteins containing soluble cCPE fragment but no transmembrane proteins. During our research on the iGEM parts we didn't find any brick about Clostridium perfringens enterotoxin (CPE). Therefore, we create our own fusion protein.
- This New Basic Part is a fusion protein composed of a reporter protein, MeosFast1, which is a modified GFP. Its composition also includes the transmembrane domain of the protein, Lamp2B, the cCPE fragment, a linker and a histidine tag.

Figure 1: Simplified scheme of the fusion protein
- As you can see the design of this part has been important for the good realization of our project.
- However, this new part is also important and interesting for the iGEM community, indeed it's a new transmembrane protein in the part registry. Claudine-4, being a protein involved in the tight junctions, can be found in all epithelial tissue in the human body and then our fusion protein could be used in various medical applications.
Design of the part
The design of this part took place in different steps :
- First we reviewed separately the different blocks of our protein in the literature and in iGEM Wikis.
- The main objective was to create a transmembrane protein that we can find on the membrane of the extracellular vesicles that's why we chose Lamp2B as anchor protein.
- References: Team:NJU-China/Parts - 2020.igem.org | UM-Macau - iGEM 2024
- However in this different wikis the teams used the entire Lamp2B whereas we have chosen to use only the transmembrane domain.
- The second important block was the cCPE fragment, which is a part of a toxin. However, we only use the non-toxic C-terminal domain (cCPE, residues 194-319), which retains Claudin-4 binding activity but lacks the N-terminal residues responsible for cytolysis.
- cCPE (residues 194-319) does not induce pore formation or cell death, as shown in literature.
-
[1]
Gao Z, McClane BA. Use of Clostridium perfringens Enterotoxin and the Enterotoxin Receptor-Binding Domain (C-CPE) for Cancer Treatment: Opportunities and Challenges.https://pmc.ncbi.nlm.nih.gov/articles/PMC3173885/#B2 "this C-terminal CPE fragment named C-CPE was nontoxic since it lacks the N-terminal regions necessary for CH-1 formation and insertion of CH-1 into membranes to form a pore"
J Toxicol. 2012;2012:981626. doi: 10.1155/2012/981626. Epub 2011 Sep 15. PMID: 21941545; PMCID: PMC3173885. -
[2]
Kokai-Kun JF, Benton K, Wieckowski EU, McClane BA. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect Immun. 1999 Nov;67(11):5634-41. doi: 10.1128/IAI. 67.11.5634-5641.1999. PMID: 10531210; PMCID: PMC96936.https://pmc.ncbi.nlm.nih.gov/articles/PMC96936 "receptor binding activity has been mapped to the extreme C terminus of the CPE protein, and it was shown that C-terminal CPE fragments, such as CPE171-319 (a CPE fragment consisting of residues 171 to 319), are nontoxic"
- It may increase drug permeability, which is an intended effect but must be handled responsibly. Moreover, there isn't evidence of pathogenicity or horizontal transfer risks associated with this domain. We will manage any risks as follows: only synthetic, codon-optimized sequences of the cCPE domain will be used, corresponding strictly to residues 194-319.
-
The third main block of the fusion protein was the reporter protein, we chose to use a
modified GFP,
mEosFast1
[3]
Maity A, Glushonkov O, Ayala I, Tacnet P, Wulffelé J, Frachet P, Brutscher B, Bourgeois D, Adam V. Decoding mEos4b day-long maturation and engineering fast-maturing variants. Protein Sci. 2025 Aug;34(8):e70234. doi: 10.1002/pro.70234. PMID: 40671276; PMCID: PMC12267110.whose sequence was provided by a team at IBS (Structural Biology Institute). This protein can be used in PALM microscopy, and the aim was to obtain high-resolution images in order to determine more precisely the localization of our protein. Unfortunately, we were unable to carry out this manipulation, but we hope to be able to do it later, or that another team interested in this fusion protein will be able to do so.
🔗 Voir la source sur PubMed
Acquisition of the part
- At the beginning of the project, the idea was to check if different constructions based on the different blocks cited upper had an impact on the liaison between the target, Claudine 4, and the ligand, cCPE.
With this in mind, we imagined the following constructions :
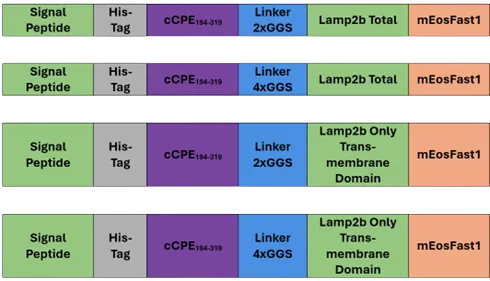
Figure 2: Simplified scheme of the four constructions we imagined
- We decided to make cloning with the restriction's sites HindIII and XbaI, even though it is an illegal site, compared with the plasmids available in our laboratory. But we submit our new part with a compatible restriction site BioBrick Standard (RFC10).
- Due to some problems in the ligation protocol, we are not able to build these four constructions so we decided to order one of them, by DNA synthesis, to continue experimenting, Consequently, we have only been able to characterize one of these four constructions:

Figure 3: Simplified scheme of fourth construction we chose
- Thanks to Pymol modeling of the interaction of our four potential fusion proteins with Claudin-4, we decided to bet on the construction shown above.
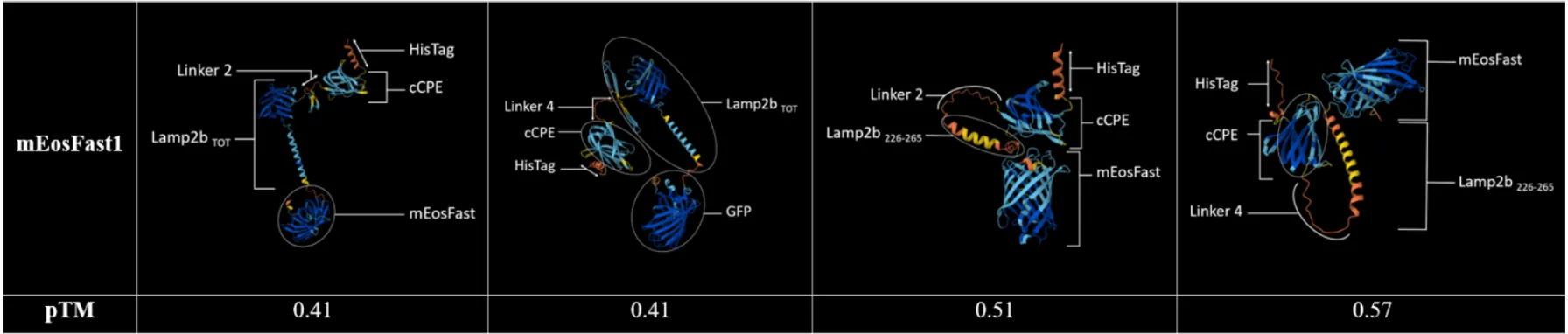
Table 1: Modelisation of the interaction between Claudin-4 and our four fusion proteins
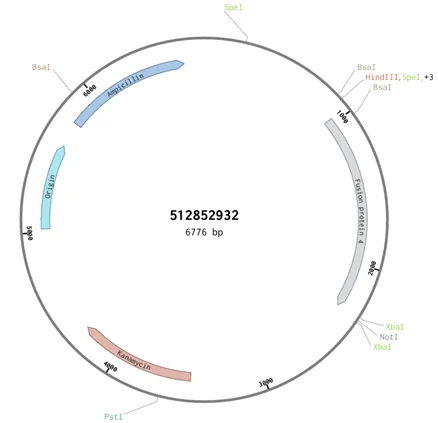
Figure 4: Scheme of the plasmide pEXP- IDT-lamp2b.trunc-linker4-cCPE ordered on IDT
Biosafety
- For use of this biobrick, a BSL-1 laboratory is sufficient. Yes, we are using a fragment of a known toxin (Clostridium perfringens enterotoxin), but only its c-term.
Recommandations
- In our case, we transformed E.coli NEB5 bacteria (from NEB) to obtain a large quantity of plasmid, which we then transfected into adherent HEK293 cells.
Experiments and controls
- Once we had received the IDT plasmid containing the gene for our lamp2b.trunc-linker4-cCPE fusion protein, we transformed it into E.coli and characterized it by agarose gel. We then transfected HEK293 cells to characterize our protein.
1) Verification of the length of the gene
- The expected length of the gene is 1,345bp, the length of the entire plasmid is 6,776bp and the length of the backbone is 5,431bp.
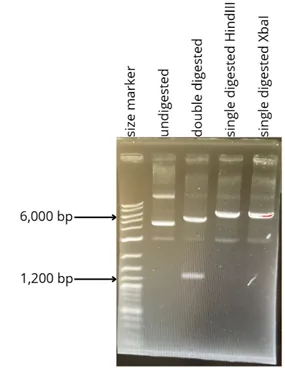
Figure 5: Agarose gel 1% showing the different parts of the plasmid after digestion or not by the enzymes HindIII and XbaI
2) Verification of expressed protein size
- We used a gel at 12% of acrylamide to separate proteins between 10 and 120 kDa. We calculated the theoretical molecular weight with the Expasy site and the expected theoretical weight of our protein is 111 kDa. We made a SDS-PAGE electrophoresis with 12% gel, then we transferred by Western Blot.
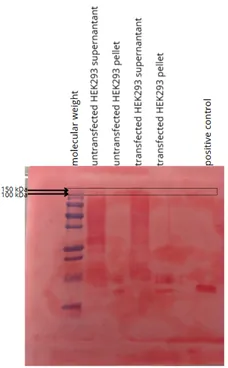
Figure 6: Western blot membrane stained with Ponceau Red to validate protein transfer
3) Transfection's results
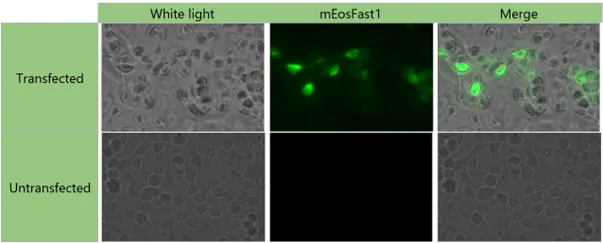
Table 2: Results of transfection with the plasmid pEXP-IDT-lamb2b.trunc-linker4
- During our transfection tests, we observed fluorescence in transfected cells, unlike in non-transfected cells. This observation confirms our belief that the fusion protein is indeed expressed in transfected cells. However, additional validation experiments, particularly regarding its functionality, are necessary in order to characterize it as accurately as possible. Unfortunately, we did not have time to carry these out.
Troubleshooting of the Western Blot
- We therefore set about establishing a protocol specific to this protein.
- First of all, we tried several different revelation methods, as the very first western blots did not show any visible bands. We therefore switched from immunorevelation using precipitation method to chemiluminescent revelation ECL (Enhanced chemiluminescence).
- Then, our transfection rates being between 30% and 50% , we concentrated the cells used for Western blotting 3 times, in order to concentrate the fusion protein as well.
- Finally, we tried out several protocols for Western blot membrane labeling. Unfortunately, none of our attempts have yet been successful.
Below is a list of parameters that can be modified to determine an effective Western blot protocol for this particular protein:
- Physical conditions of membrane transfer: time, voltage, amperage. As can be seen on the Ponceau Red-stained Western Blot membrane, the bands for high molecular weights are barely visible. Perhaps the protein of interest was not transferred in our tests.
- Better positive control, in our case the molecular weight of the positive control was very different from the theoretical molecular weight of the protein of interest. As a result, we used a percentage gel covering a wide range of molecular weights, which was not ideal for either the control or the protein of interest.
- Testing other types of cell lysis. The lysis we used was with RIPA buffer, but it is possible to choose another type: detergent lysis, sonication, Laemmli buffer lysis...