



Overview
In the AGEs Robber project, sRAGE or the N-glycan depleted sRAGE are fused with collagen
binding domain (CBD), enabling its immobilization on the collagen layer of AGEs-Thwart
Patch (see Design for details).
In the beginning, sRAGE and CBD were connected via a linker. The brainstorming further
brings the idea of switchable molecule by intein splicing system. Therefore, the linker is
replaced by Npu DnaE split intein.
Whether the initial fusion protein system or the subsequent switchable intein splicing
system, the propre protein folding is essential for protein function. Accordingly, we
performed structural modeling of sRAGE-CBD or switchable sRAGE-CBD fusion protein by
AlphaFold3.
However, AlphaFold3 assigned relatively low confidence scores to the inter-domain
connecting regions, due to lack of homologous structure in training databases. To
further evaluate the reliability of predicted structure, a Ramachandran plot was generated
by PyMOL to examine the distribution of backbone dihedral angles, confirming the absence
of significantly unreasonable conformations.
Preparation for Protein Modeling
Sequences
The first step of modeling is collecting the protein sequences. In our project, the
protein sequences are listed below:
-
Protein sequences
sRAGE AQNITARIGE PLVLKCKGAP KKPPQRLEWK LNTGRTEAWK VLSPQGGGPW
DSVARVLPNG SLFLPAVGIQ DEGIFRCQAM NRNGKETKSN YRVRVYQIPG
KPEIVDSASE LTAGVPNKVG TCVSEGSYPA GTLSWHLDGK PLVPNEKGVS
VKEQTRRHPE TGLFTLQSEL MVTPARGGDP RPTFSCSFSP GLPRHRALRT
APIQPRVWEP VPLEEVQLVV EPEGGAVAPG GTVTLTCEVP AQPSPQIHWM
KDGVPLPLPP SPVLILPEIG PQDQGTYSCV ATHSSHGPQE SRAVSISIIE PMutated sRAGE AQQITARIGE PLVLKCKGAP KKPPQRLEWK LNTGRTEAWK VLSPQGGGPW
DSVARVLPQG SLFLPAVGIQ DEGIFRCQAM NRNGKETKSN YRVRVYQIPG
KPEIVDSASE LTAGVPNKVG TCVSEGSYPA GTLSWHLDGK PLVPNEKGVS
VKEQTRRHPE TGLFTLQSEL MVTPARGGDP RPTFSCSFSP GLPRHRALRT
APIQPRVWEP VPLEEVQLVV EPEGGAVAPG GTVTLTCEVP AQPSPQIHWM
KDGVPLPLPP SPVLILPEIG PQDQGTYSCV ATHSSHGPQE SRAVSISIIE P -
Linker sequences
Linkers are short peptide sequences used to connect different domains within fusion proteins. In the beginning, two types of linkers with rigid characteristics were selected for structural-modeling. Their rigidity helps minimize unwanted interactions between adjacent protein domains. (Arai et al., 2001) (Bhandari et al., 1986)
A(EAAAK)n A(n= 2) AEAAAKEAAA KA (AP)7 APAPAPAPAP APAP -
CBD sequences
Collagen-binding domain is a protein domain that specifically binds to collagen. In our project, CBD is incorporated to anchor the fusion protein onto collagen-containing hydrogels. We selected the lumican LRR 5–7 and the fibromodulin LRR5-7 region because previous studies reported their low dissociation constant (Kd), indicating high binding affinity toward collagen. (Kalamajski & Oldberg, 2009)
Lumican LRR 5-7 NLTFIHLQHN RLKEDAVSAA FKGLKSLEYL DLSFNQIARL PSGLPVSLLT
LYLDNNKISN IPDEYFKRFibromodulin LRR 5-7 NLTALYLQHN EIQEVGSSMR GLSLILLDLS YNHLRKVPDG LPSALEQLYM
EHNNVYTVPD SYFRG -
Npu DnaE split intein sequences
Inteins are self-cleaving protein elements that catalyze protein splicing, excising themselves while ligating the flanking N- and C-exteins through a native peptide bond. We selected the Npu DnaE intein system because it exhibits exceptionally fast splicing kinetics (short half-life). (Iwai, Züger, Jin, & Tam, 2006)
RGK-(Nup-DnaEN) CLSYETEILT VEYGLLPIGK IVEKRIECTV YSVDNNGNIY TQPVAQWHDR
GEQEVFEYCL EDGSLIRATK DHKFMTVDGQ MLPIDEIFER ELDLMRVDNL PN(Nup-DnaEC)-CWE MIKIATRKYL GKQNVYDIGV ERDHNFALKN GFIASN -
Enhanced Green Fluorescent Protein
Enhanced Green Fluorescent Protein (EGFP) is used as a fluorescent reporter. In our project we used EGFP to verify successful transfections in mammalian cells and to visually confirm surface attachment of our fusion proteins onto collagen hydrogels.
EGFP MVSKGEELFT GVVPILVELD GDVNGHKFSV SGEGEGDATY GKLTLKFICT
TGKLPVPWPT LVTTLTYGVQ CFSRYPDHMK QHDFFKSAMP EGYVQERTIF
FKDDGNYKTR AEVKFEGDTL VNRIELKGID FKEDGNILGH KLEYNYNSHN
VYIMADKQKN GIKVNFKIRH NIEDGSVQLA DHYQQNTPIG DGPVLLPDNH
YLSTQSALSK DPNEKRDHMV LLEFVTAAGI TLGMDELYK
Interpretation of AlphaFold Confidence Metrics
pLDDT value
Predicted Local Distance Difference Test (pLDDT) is a confidence score used by AlphaFold to indicate how reliable the predicted structure is at each amino acid residue. A higher score means AlphaFold has greater confidence in the accuracy of that region. In AlphaFold‘s colored structural models, residues are represented according to their pLDDT values:
- 90–100 (blue): Very high confidence; atomic coordinates can be trusted.
- 70–90 (cyan/green): High confidence; overall reliable with minor possible errors.
- 50–70 (yellow): Low confidence; structural prediction may be inaccurate and should be interpreted with caution.
- <50 (orange–red): Very low confidence; often indicates intrinsically disordered regions where AlphaFold cannot provide an accurate prediction.
Predicted Aligned Error plot
The Predicted Aligned Error (PAE) is an AlphaFold output that estimates the pairwise
confidence of residue positions within a predicted protein structure. Unlike pLDDT,
which evaluates individual residues, the PAE matrix describes the relative accuracy
between all residue pairs, making it particularly useful for assessing domain packing,
flexible regions, and inter-domain orientations.
In a PAE plot:
- X-axis (Scored Residue): Amino acid sequence index (residue 1 to the last).
- Y-axis (Aligned Residue): Amino acid sequence index.
- Color scale:
- Dark green (< 5 Å): High confidence in relative positioning.
- Light green to white (> 20 Å): Low confidence, indicating flexibility or uncertainty.
Build
Build the structure mutated sRAGE
We first modeled the mutated sRAGE using AlphaFold to evaluate its structural stability after mutation. Based on the pLDDT color scheme, we observed that the folding of each domain appeared to be well maintained.
- Mutated sRAGE
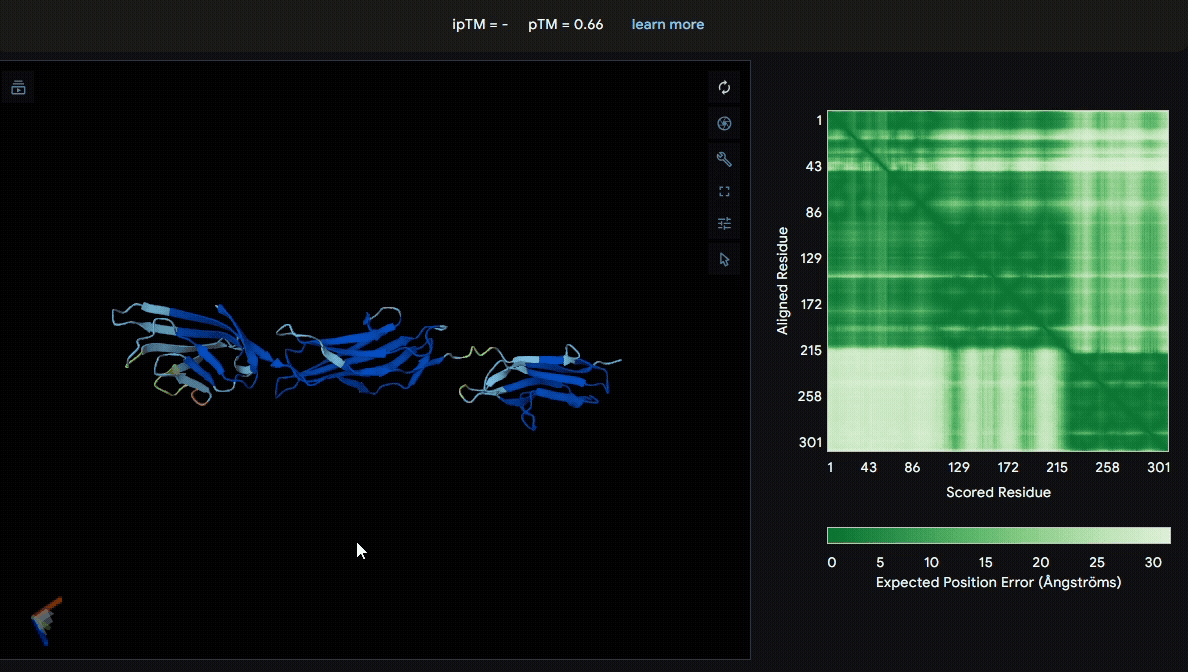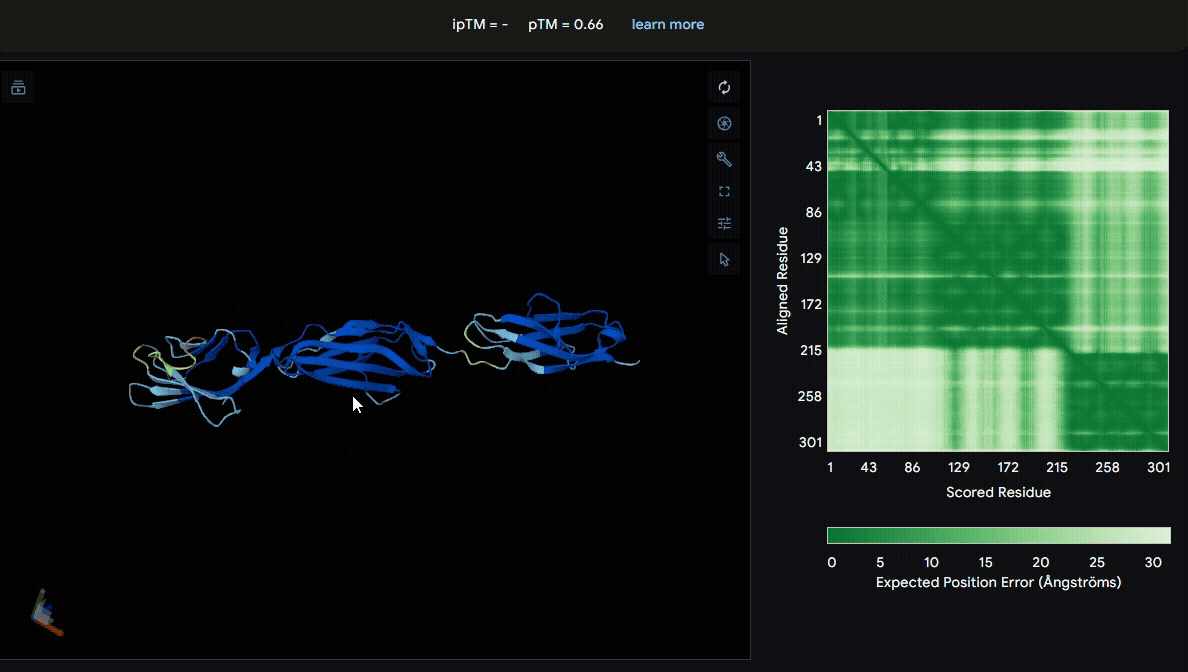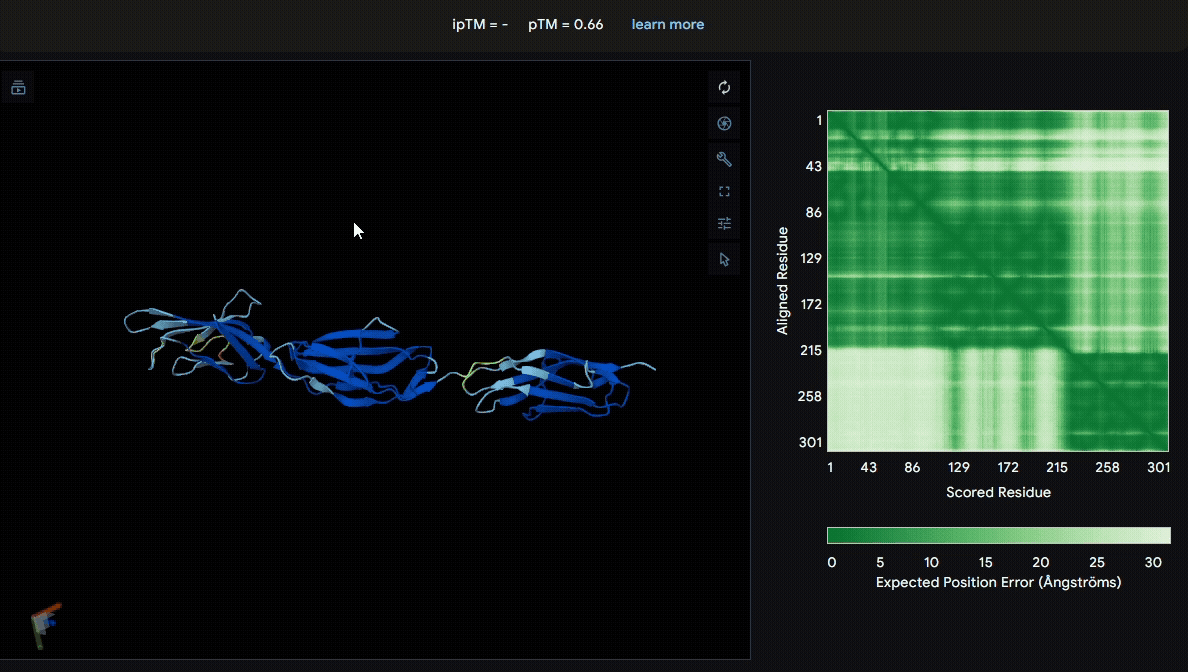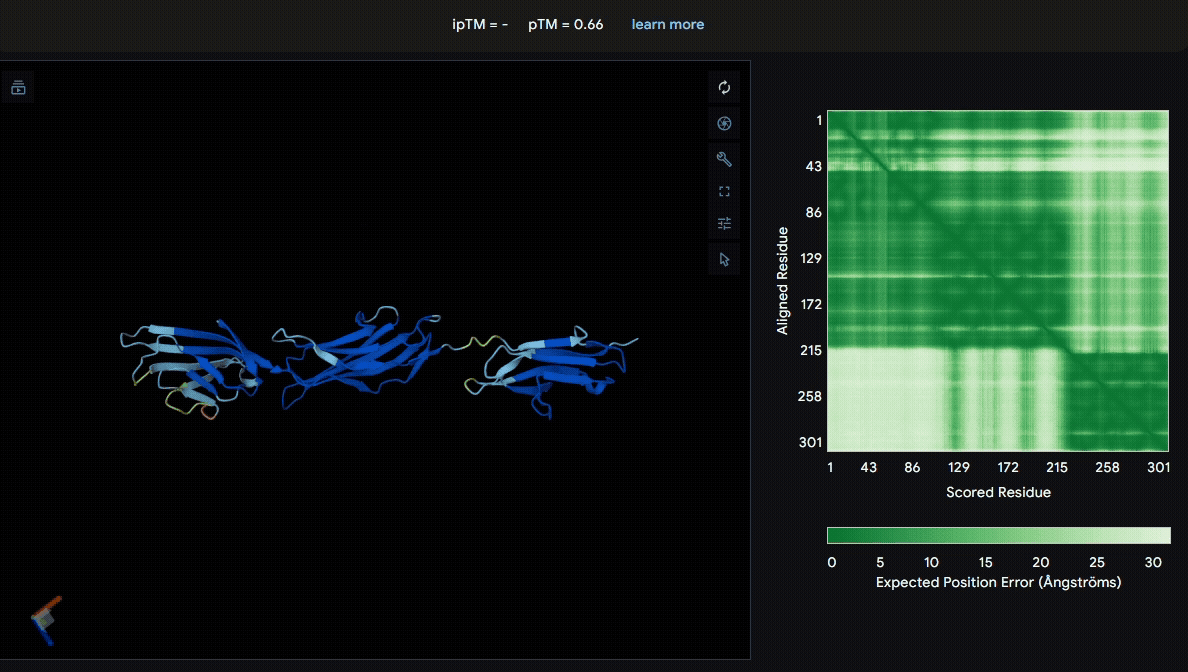
Build the structure of collagen binding domains
We used LRR5–7 domains from lumican and fibromodulin for modeling. From the pLDDT color scheme, we observed that the folding of each domain appeared to be well maintained.
- Lumican (Domain :LRR5-7)
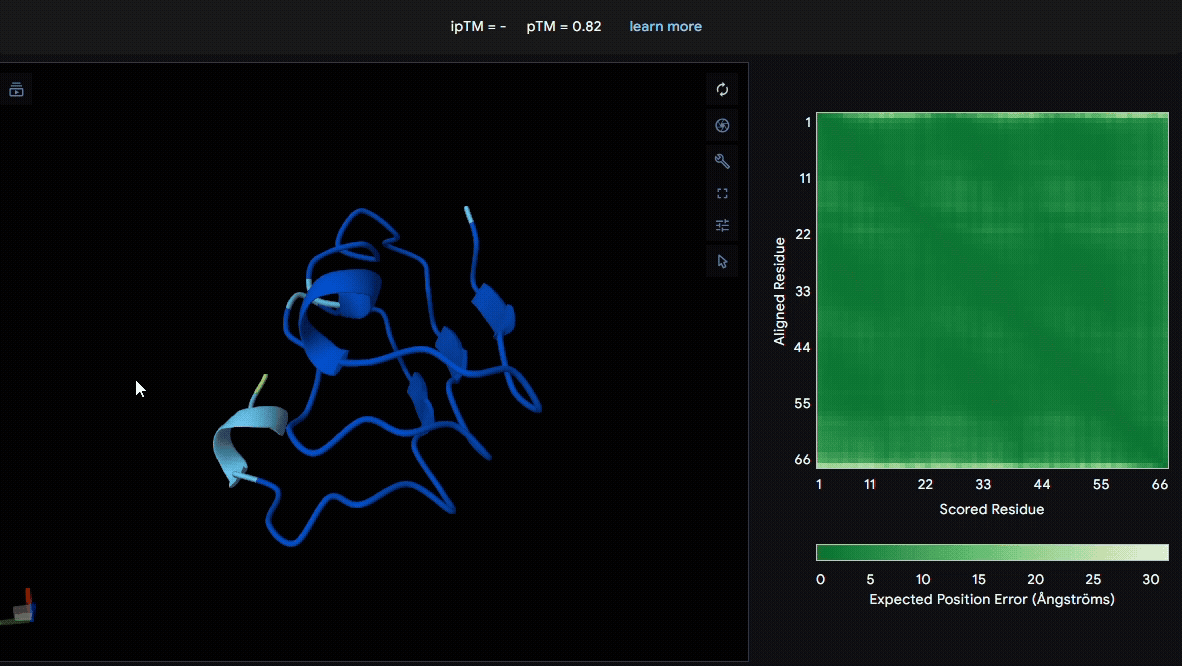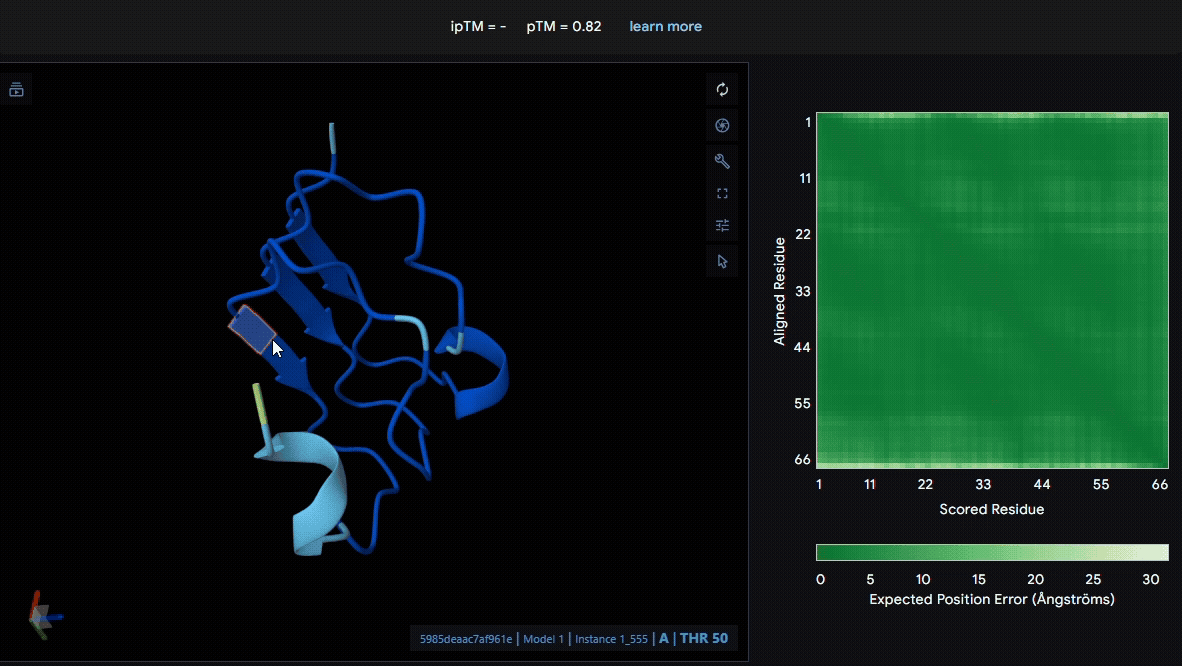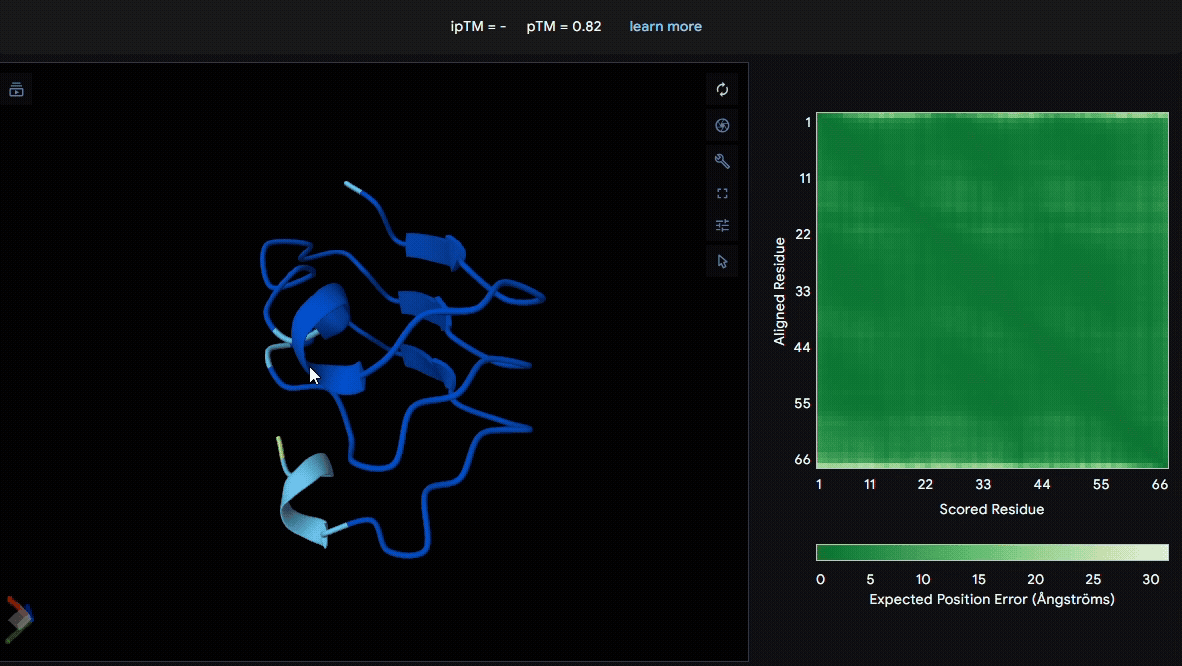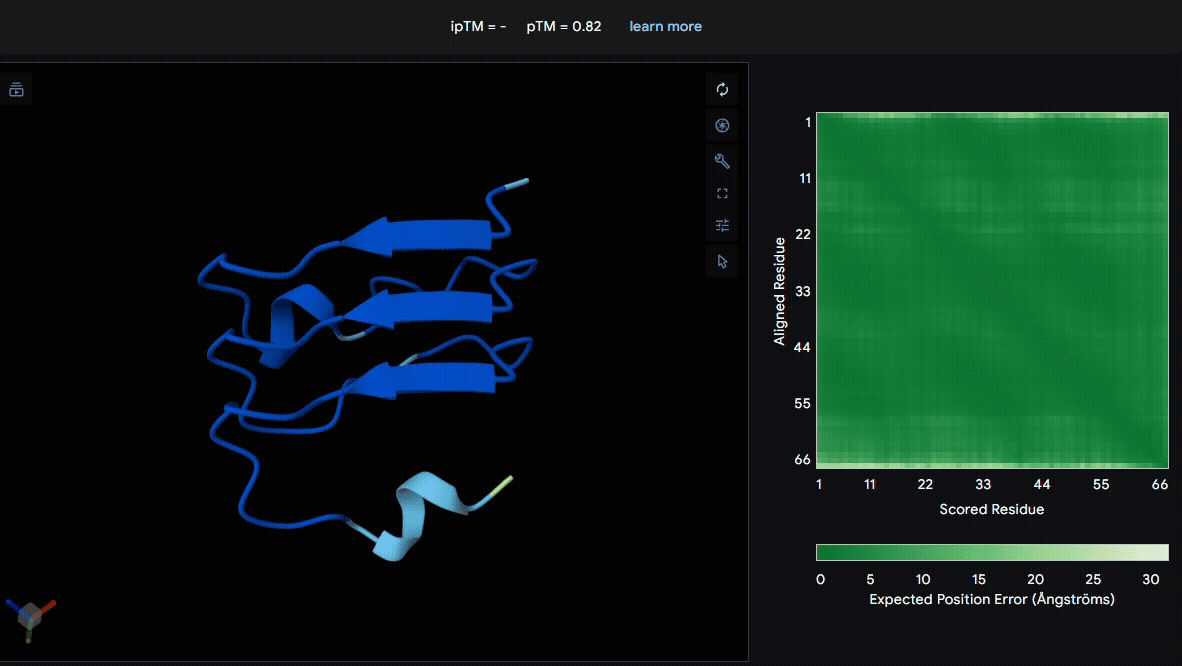
- Fibromodulin (Domain : LRR5-7)
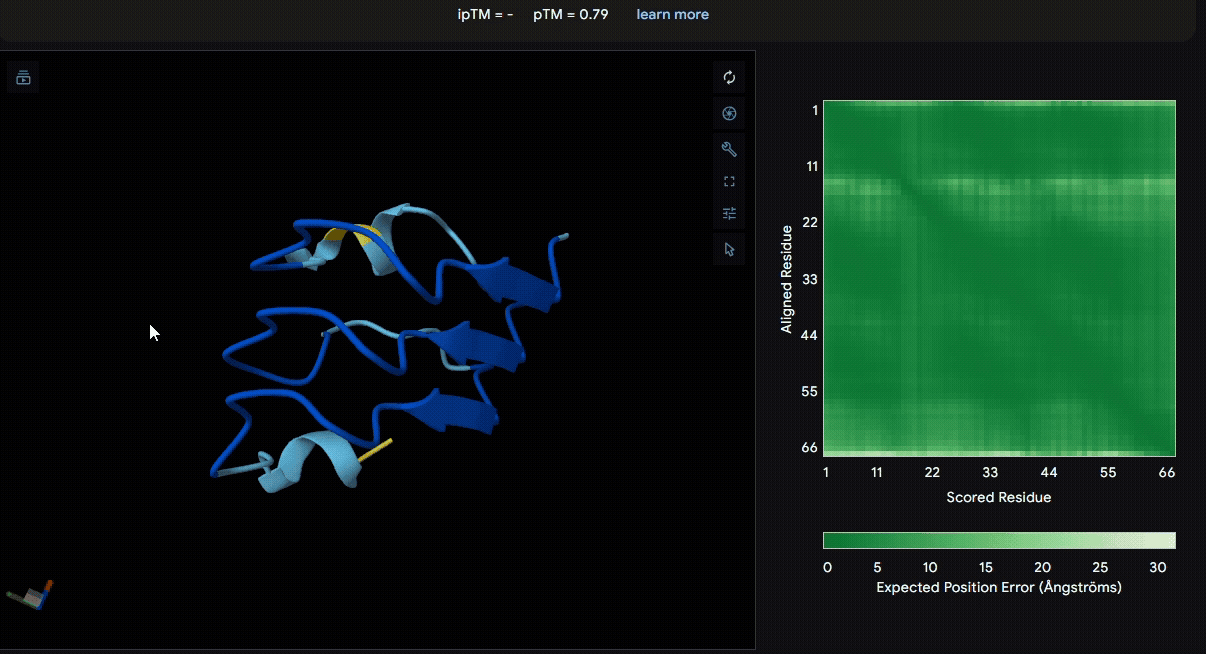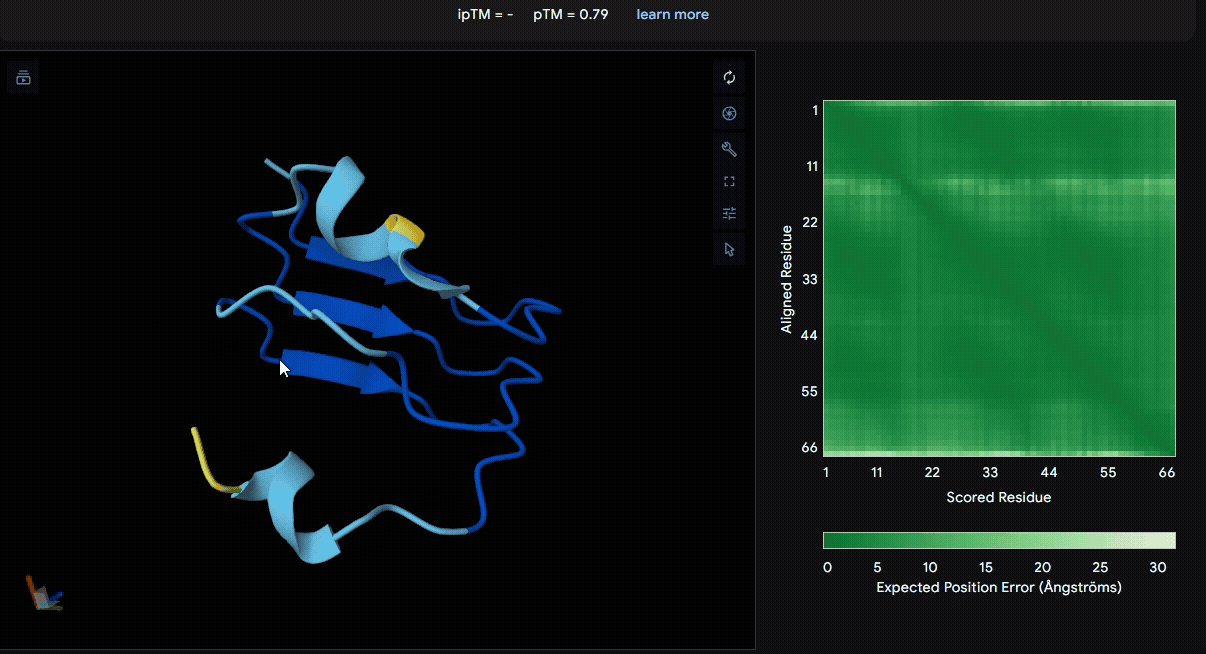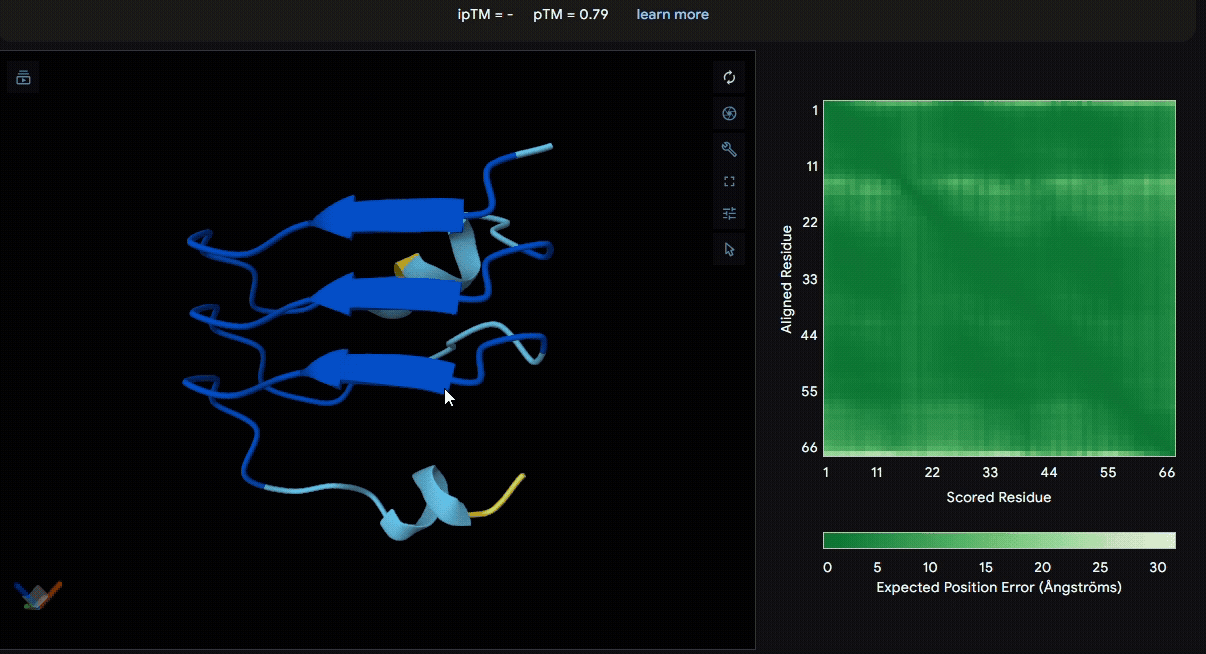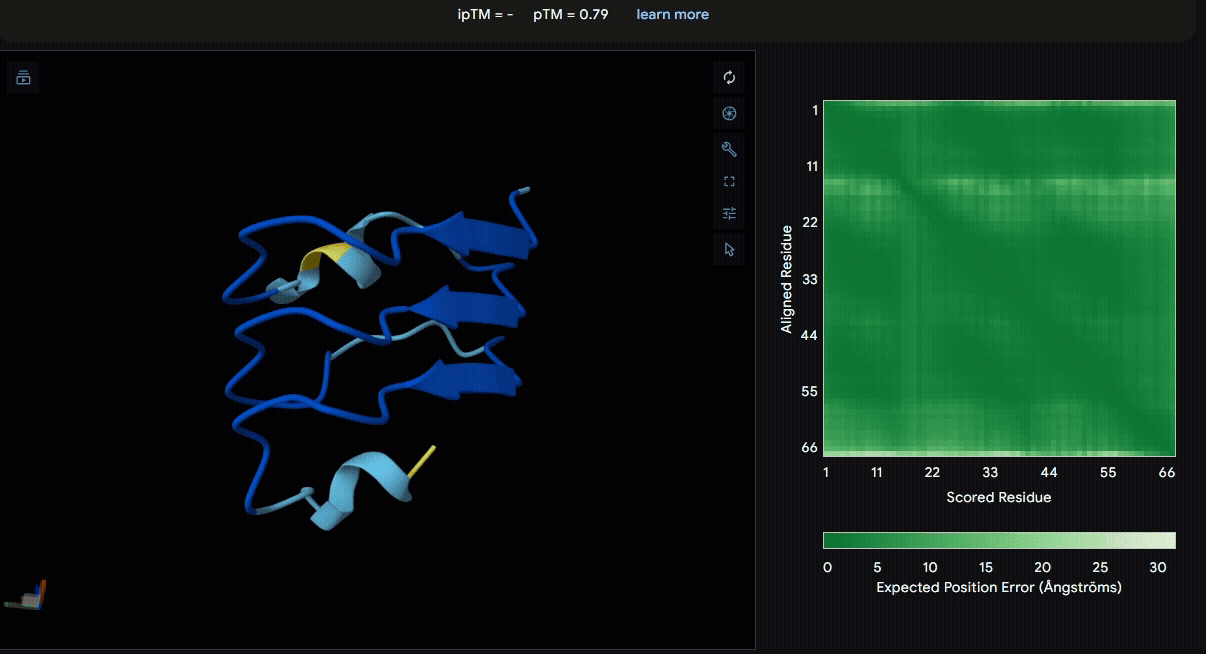
Build the fusion protein with linker
To identify a suitable CBD for our design and to determine whether a linker was required to connect CBD and sRAGE, we performed fusion protein structure prediction using AlphaFold. We tested the following combinations:(Blue word is linker; red word is CBD)
- sRAGE_No linker (EAK)_Lumican
- sRAGE_No linker (EAK)_Fibromodulin
- sRAGE_(AP)7_Lumican
- sRAGE_(AP)7_Fibromodulin
- sRAGE_A(EAAK)nA_Fm
- sRAGE_A(EAAK)nA_Lum
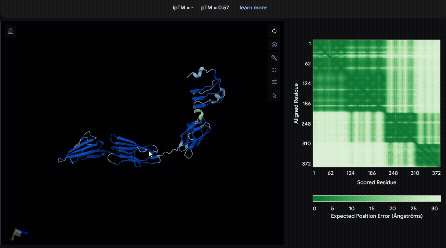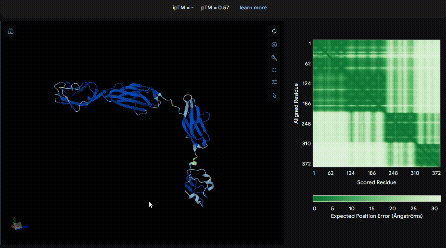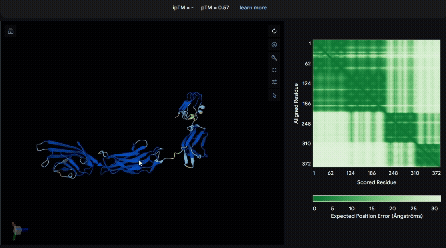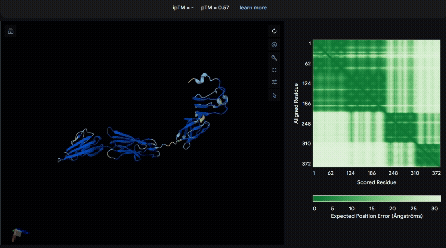
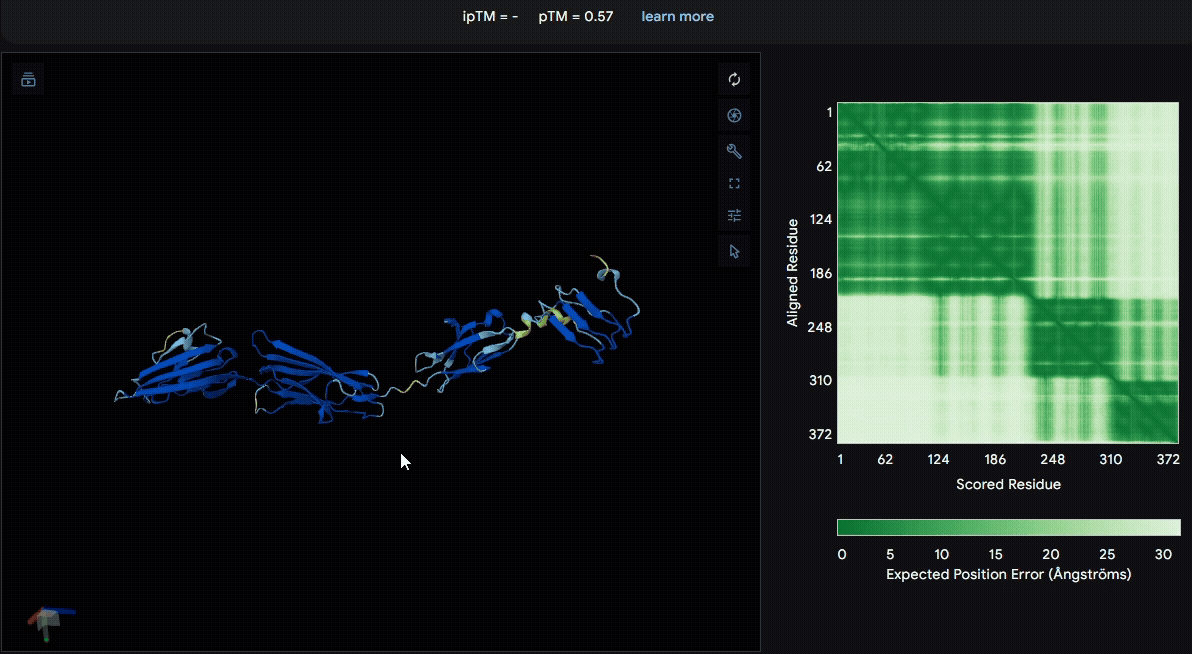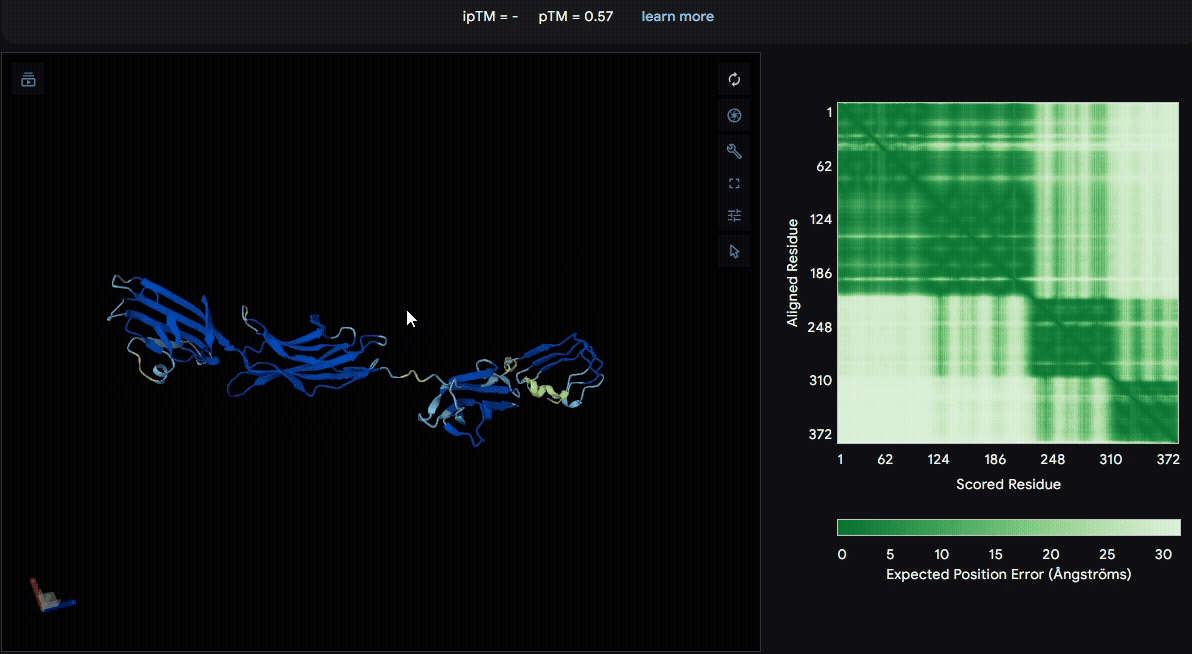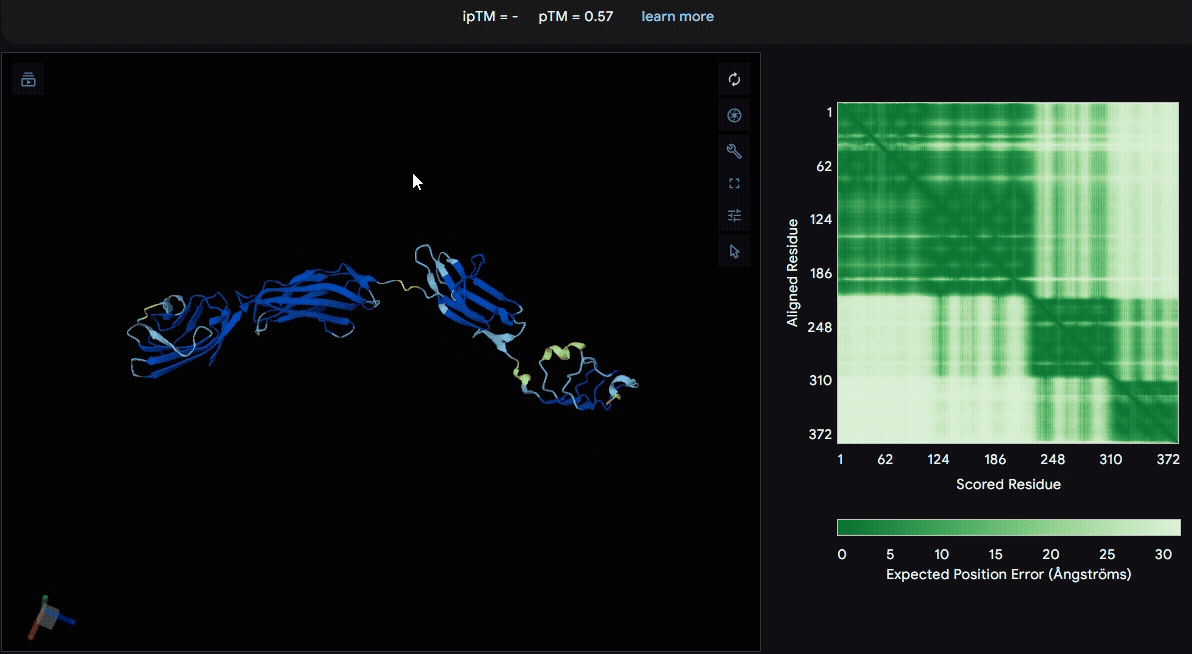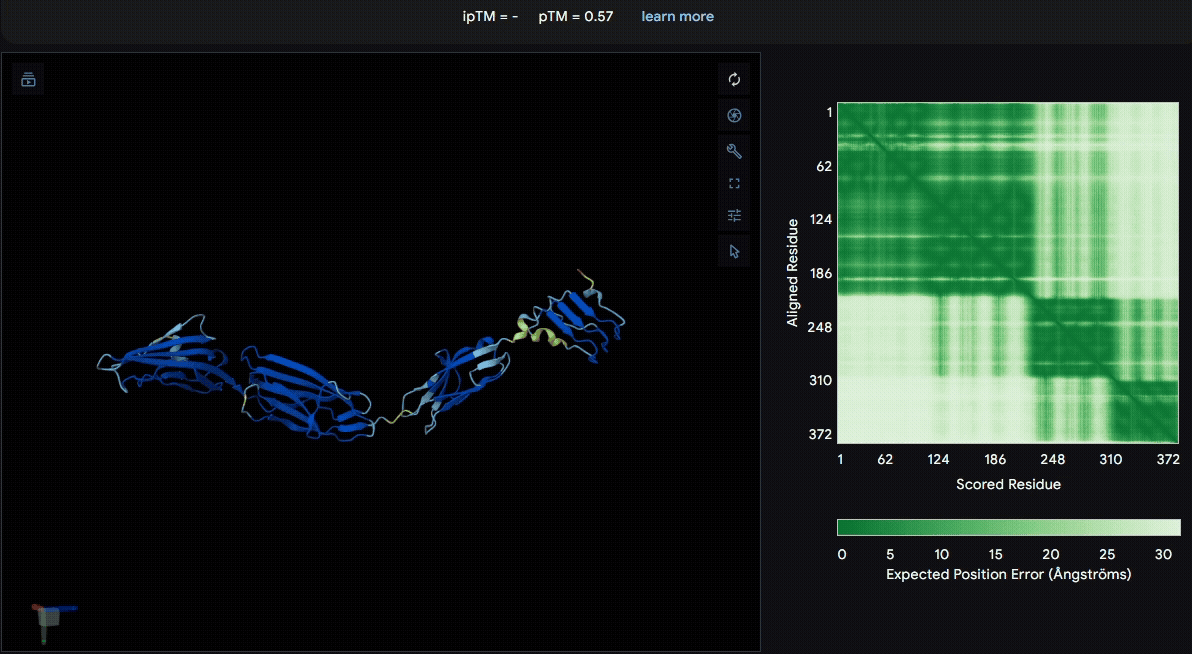
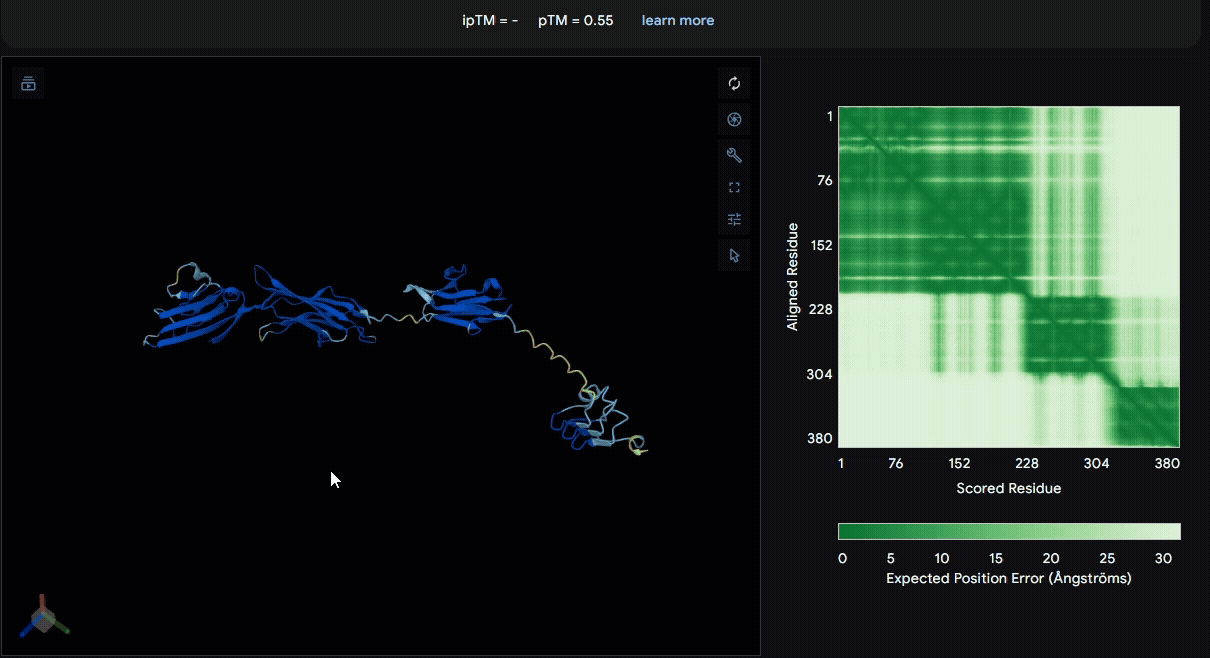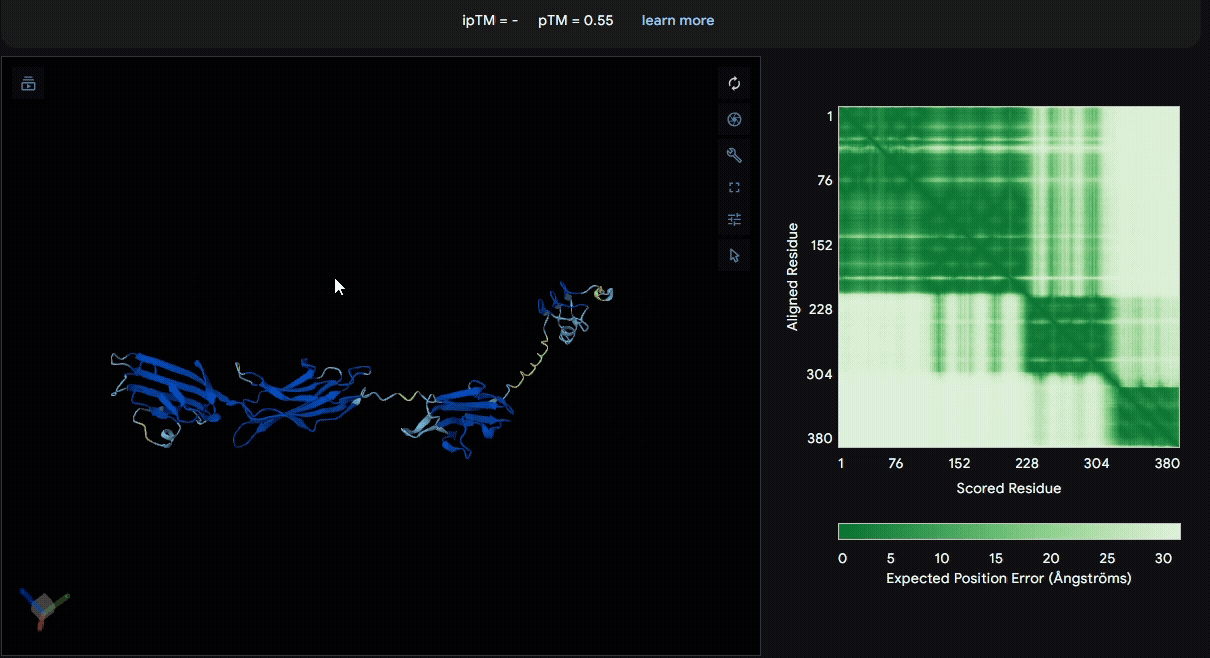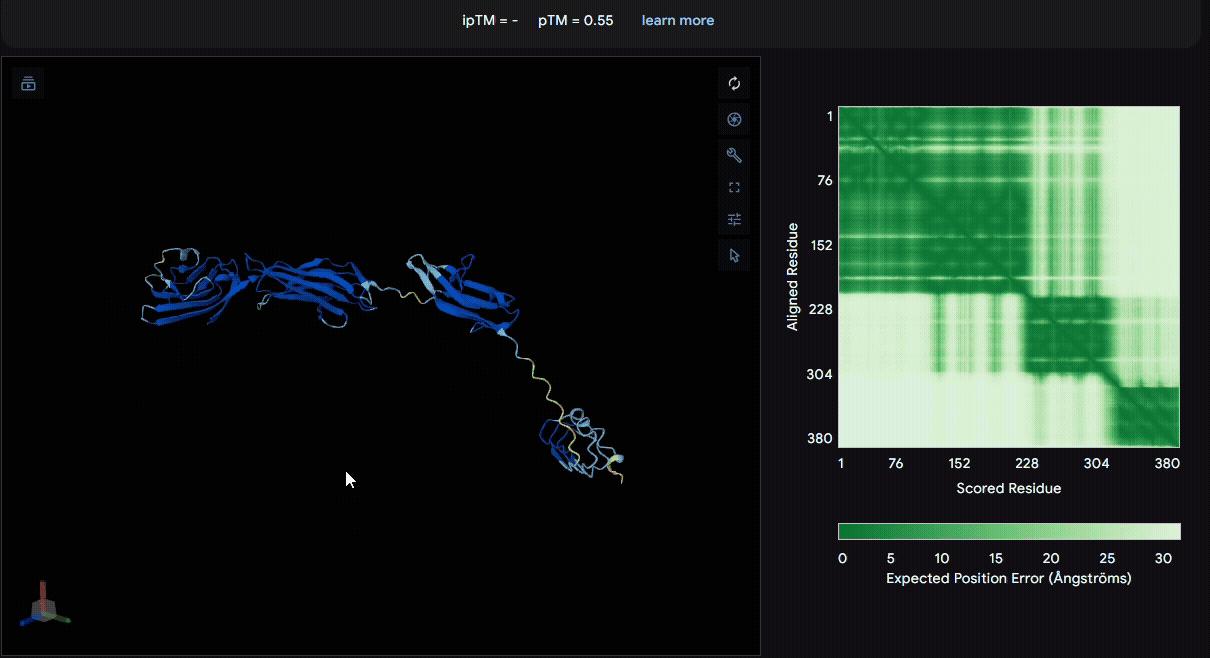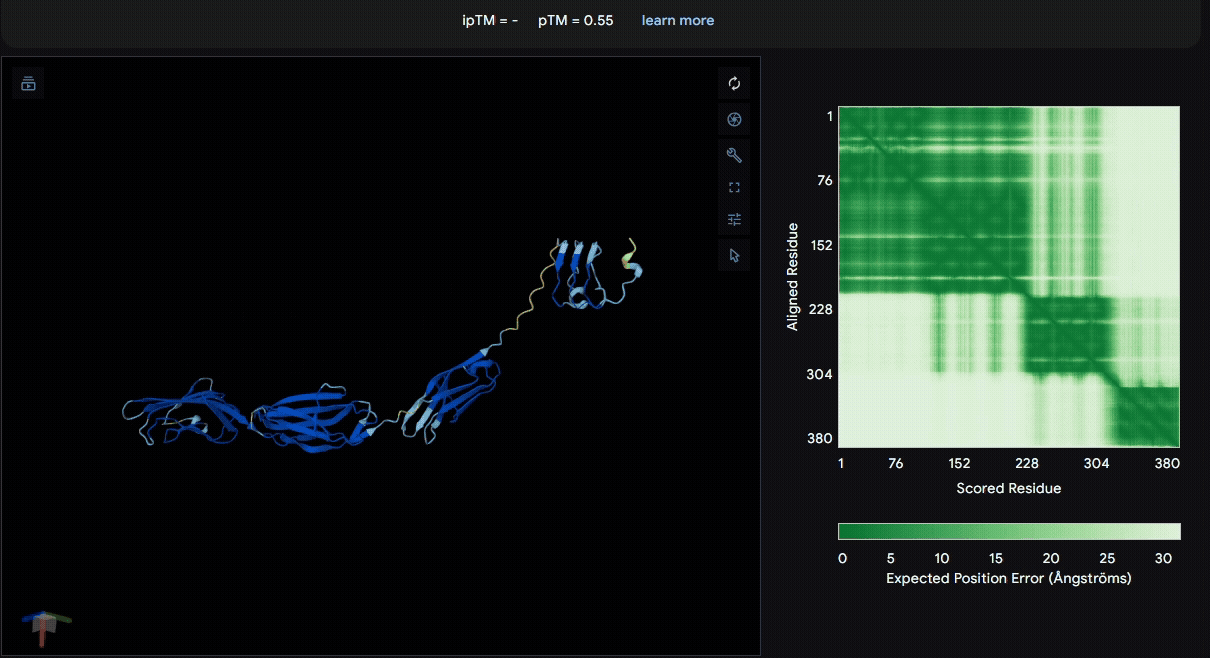
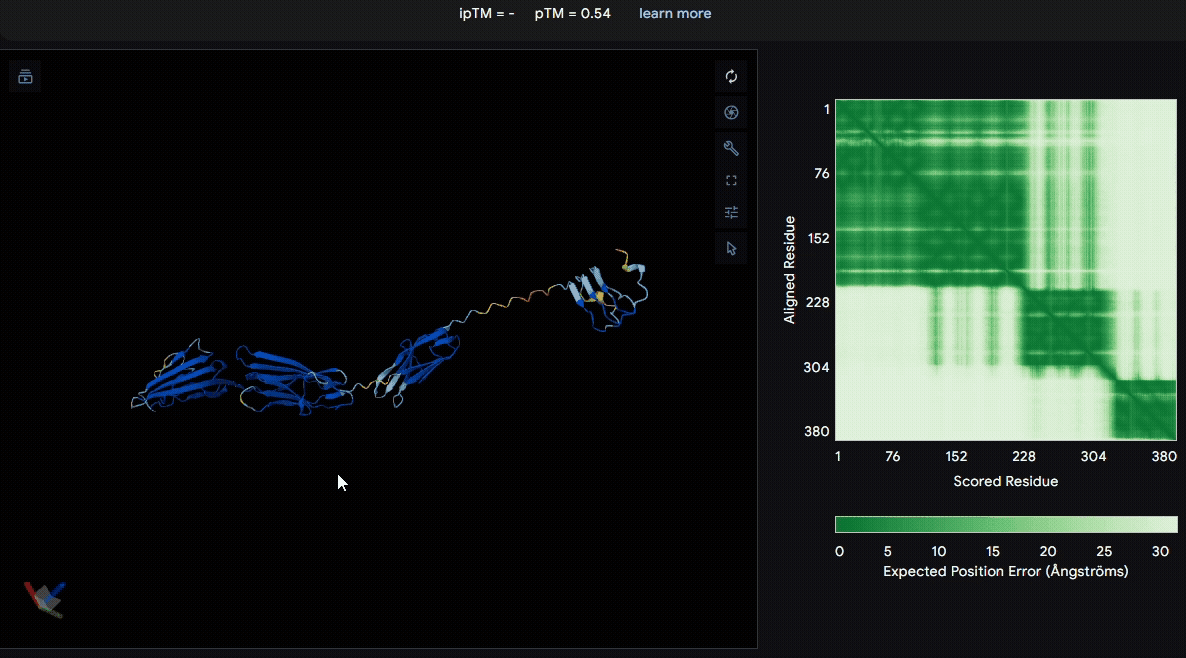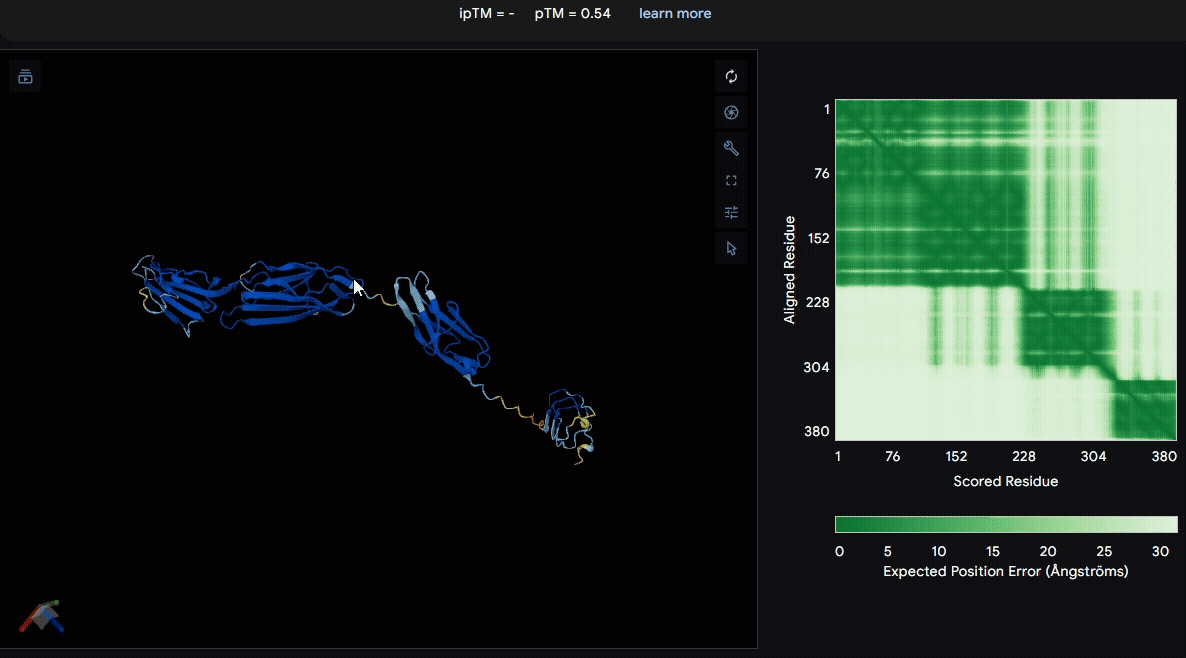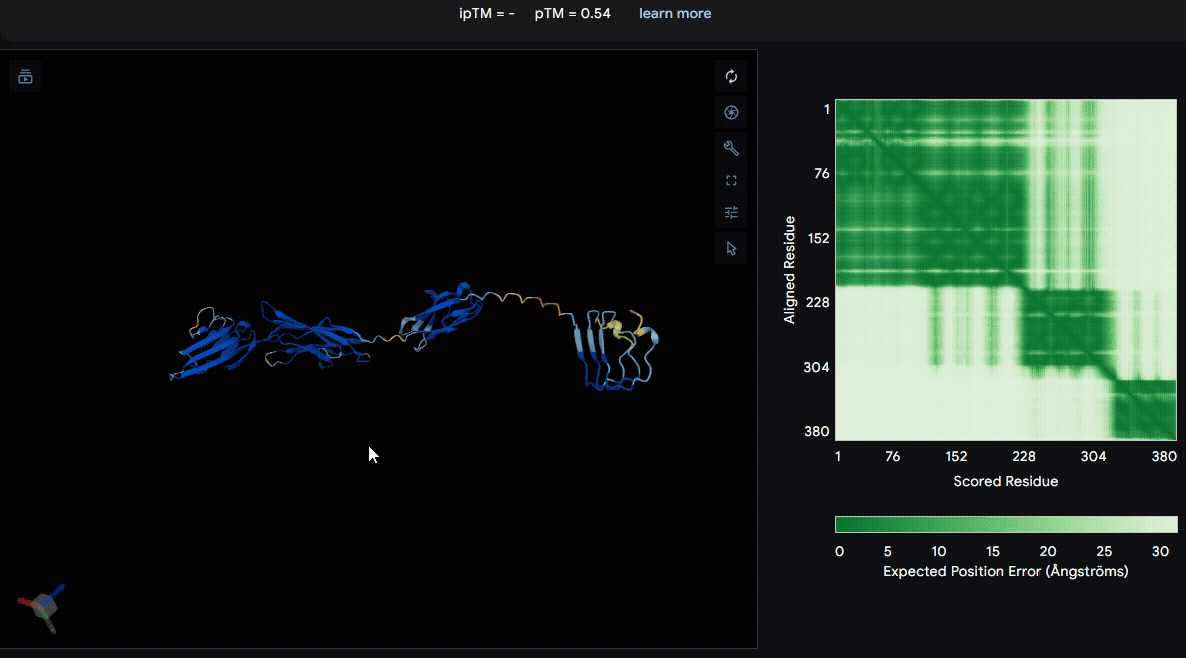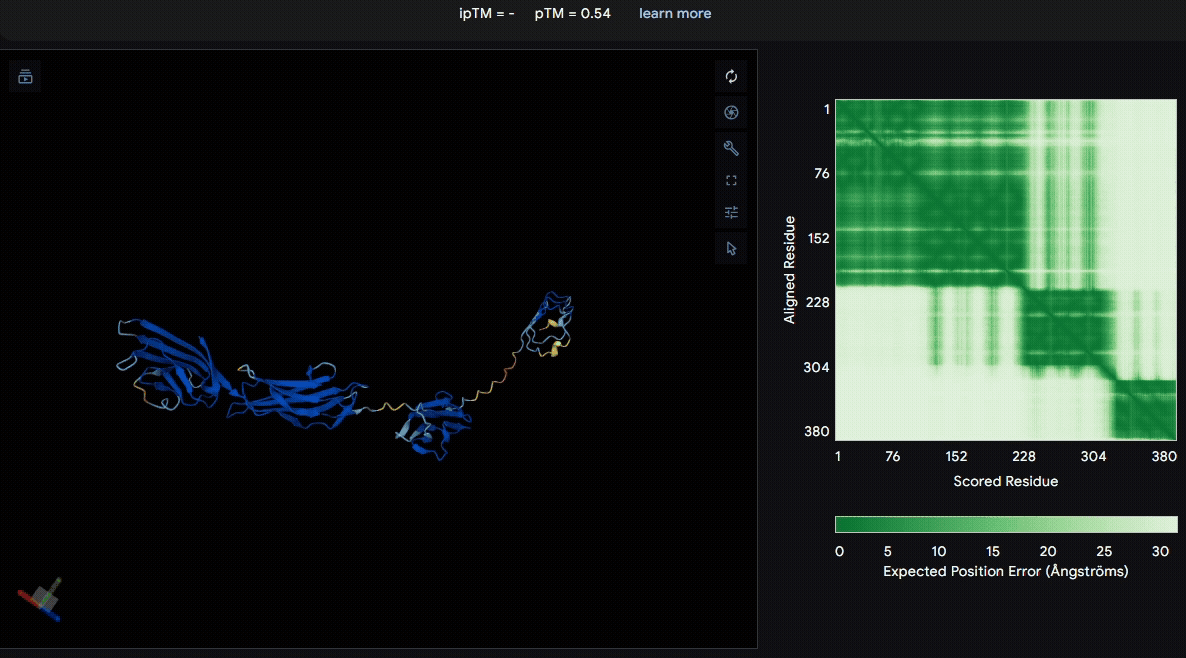
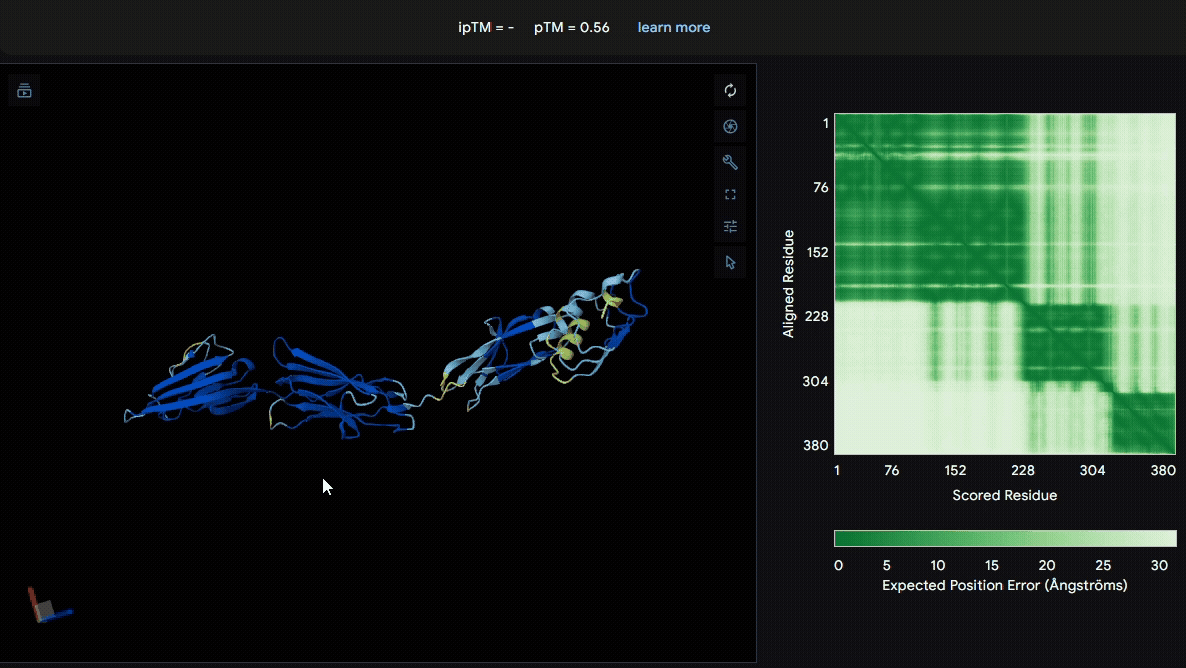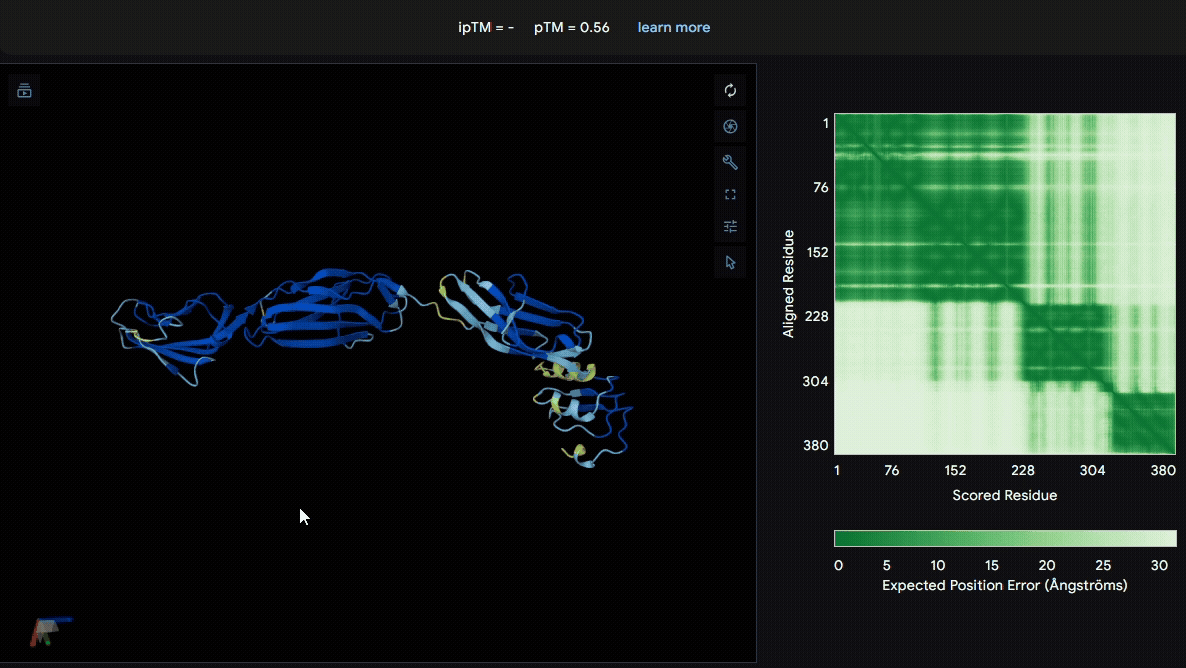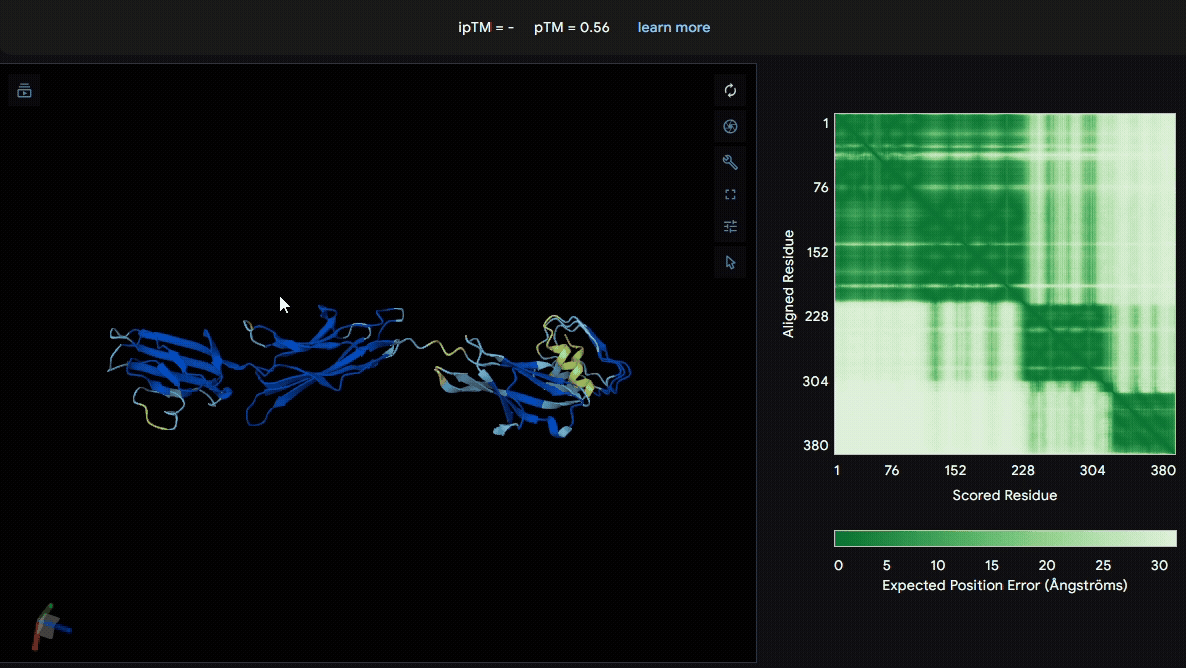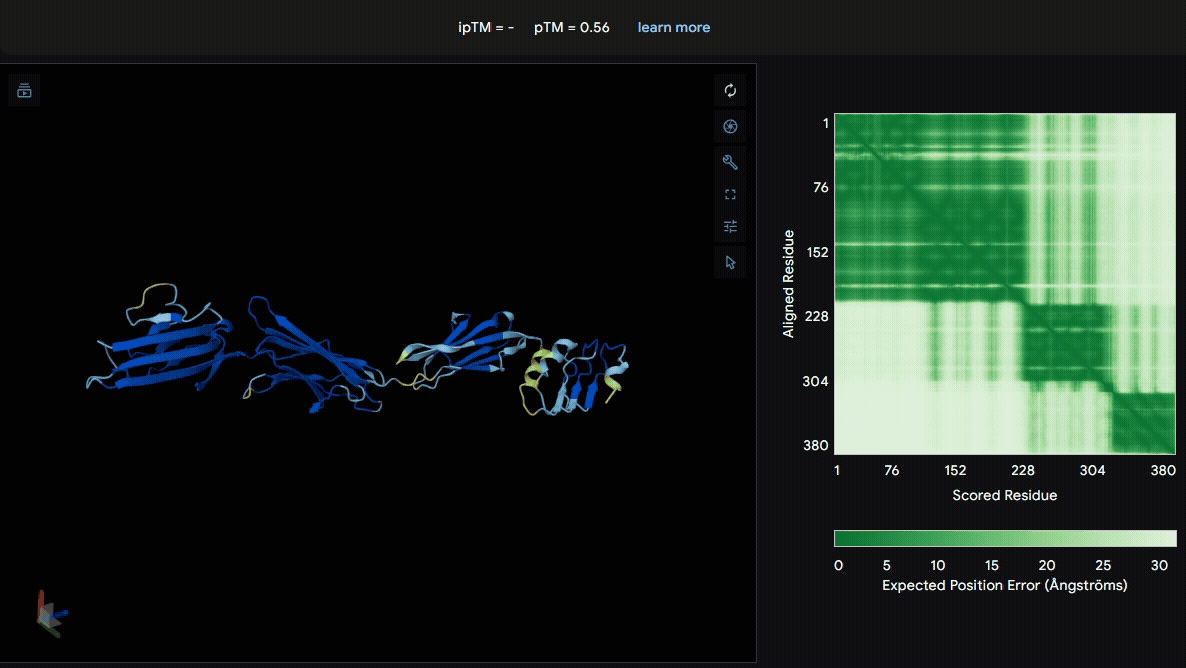
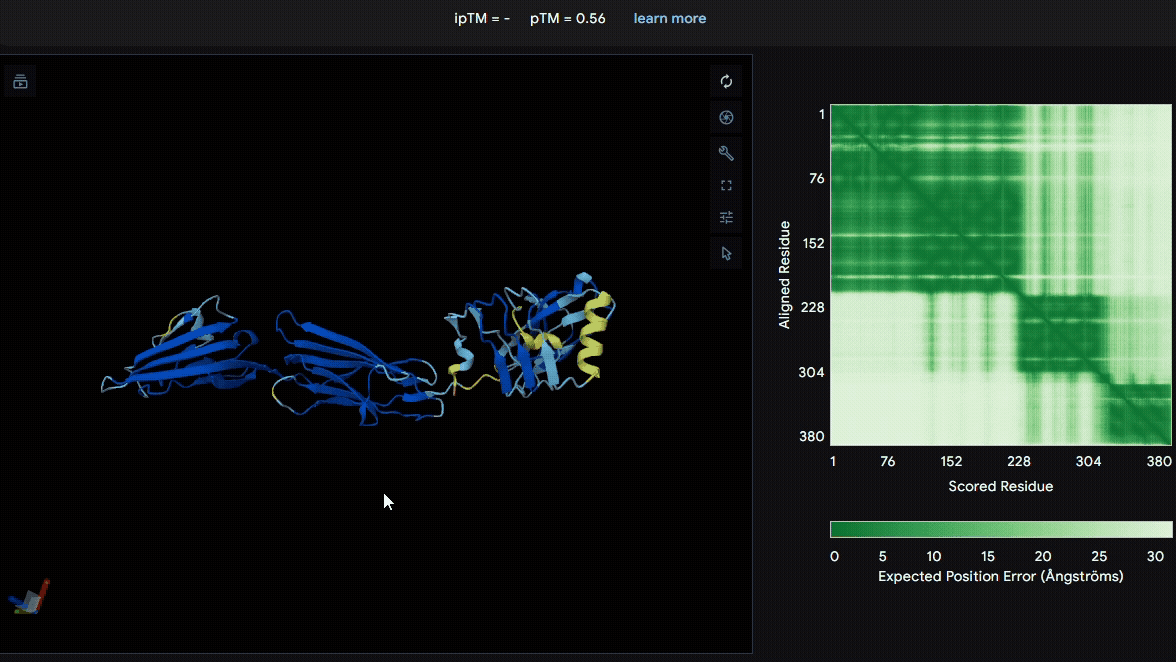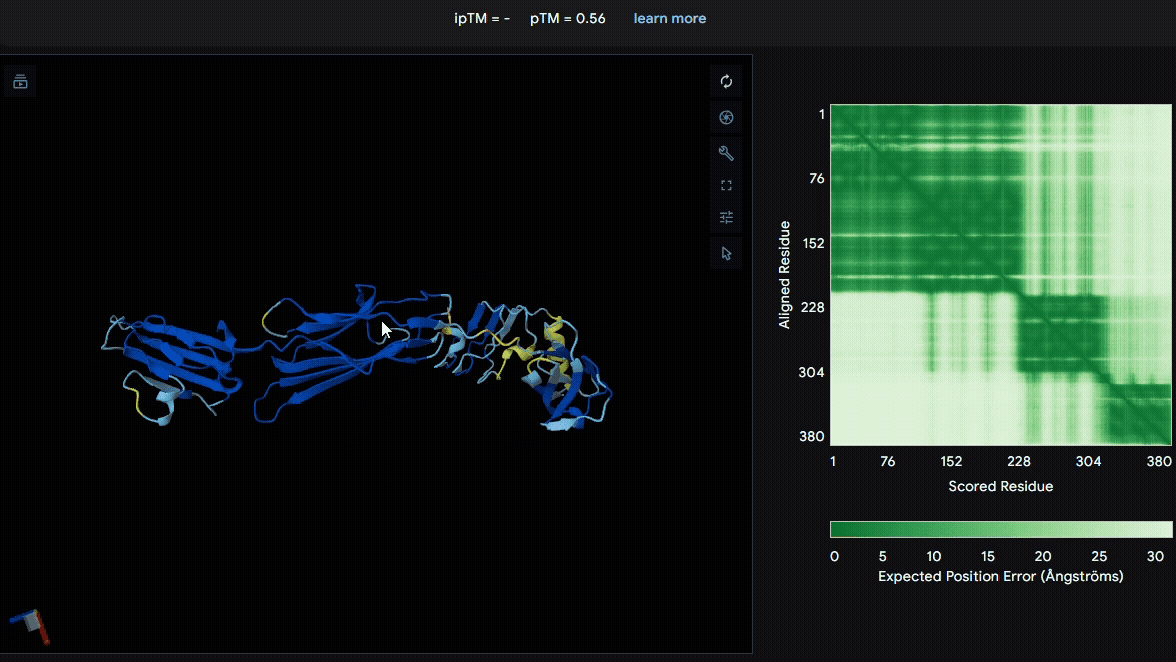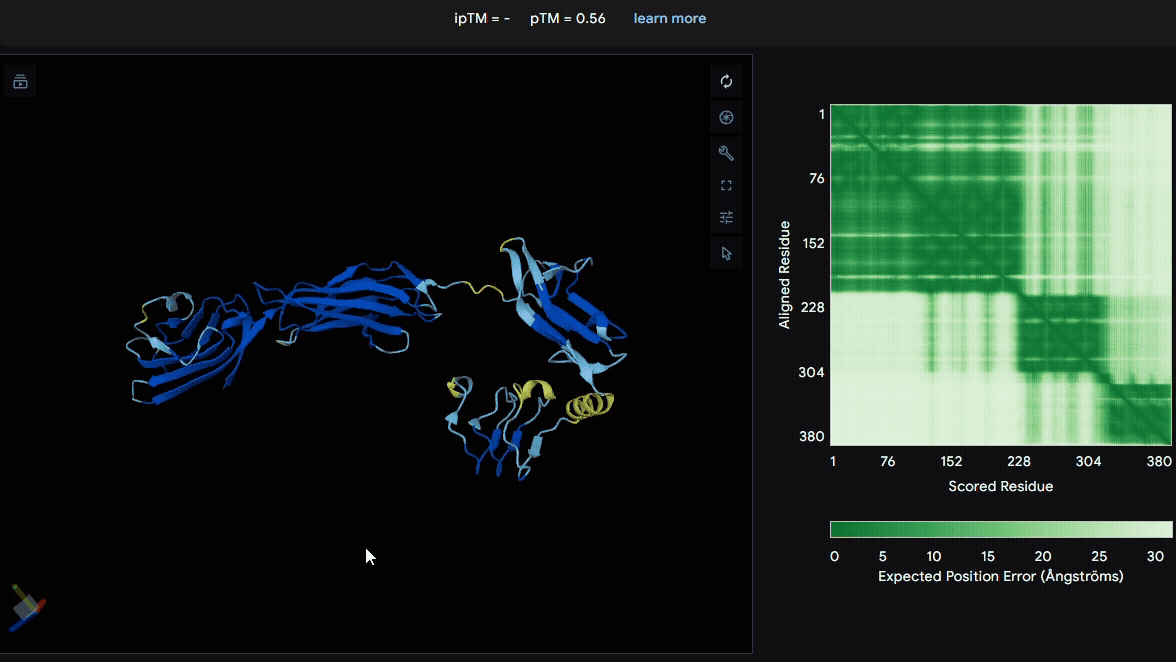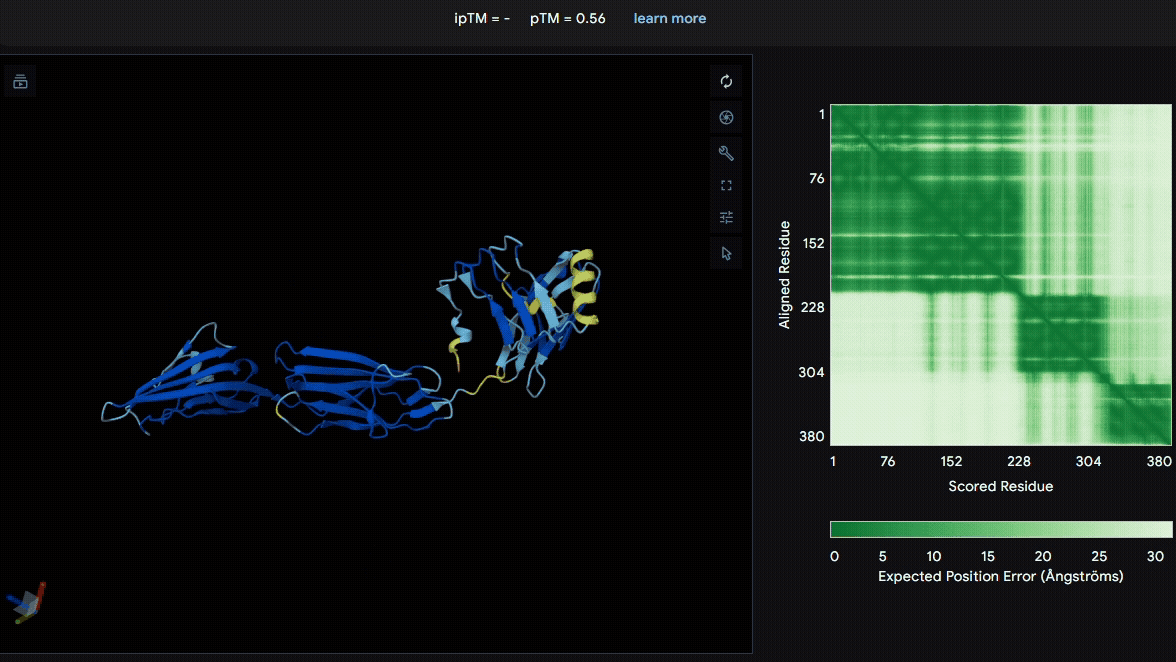
Among the six tested combinations, lumican showed better performance than fibromodulin, and the no-linker construct best met our criteria. While these comparisons allowed us to identify relatively more favorable constructs, all six models still exhibited a substantial number of white regions. To further assess the reliability of the predicted structures, Ramachandran plots of all six models were generated using PyMOL.
Model Validation through Ramachandran Plot Analysis
The Ramachandran plot is an analytical tool used to assess the stereochemical quality of
protein structures. It plots the backbone dihedral angles φ (phi) and ψ (psi) on the
x- and y-axes, respectively, with each point representing the angle combination of a
single amino acid residue. Due to steric hindrance and chemical bond constraints, only
certain regions are considered “allowed regions,” typically corresponding to α-helices,
β-sheets, and left-handed helices.
In the plot, red and yellow contours indicate the most favored and allowed dihedral
angle regions, respectively, while black dots represent the actual φ/ψ angles of residues
in the protein. If most residues fall within the allowed regions, the model is considered
structurally reasonable and stable. Conversely, a significant number of points in disallowed
regions may indicate conformational issues or prediction errors. Therefore, the Ramachandran
plot is widely used to validate protein models and serves as an important reliability
metric, particularly in molecular modeling and structure prediction studies.
To further evaluate the structural reliability of the predicted models, Ramachandran plots
were generated for all six structures.






Build the fusion protein with Npu DnaE split intein
Although previous modeling indicated that the fusion protein without a linker using CBD
from lumican is the most favorable, we adapted the patch for broader clinical applications
by introducing the Npu DnaE split intein system, enabling modular assembly of sRAGE or other
therapeutic proteins such as antimicrobial peptides with CBD. Therefore, we modeled the
following fusion protein constructs:
-
(Nup-DnaEC)-CWE-6xHis-CBD(Lumican)
A His-tag was introduced into the sequence to facilitate protein purification.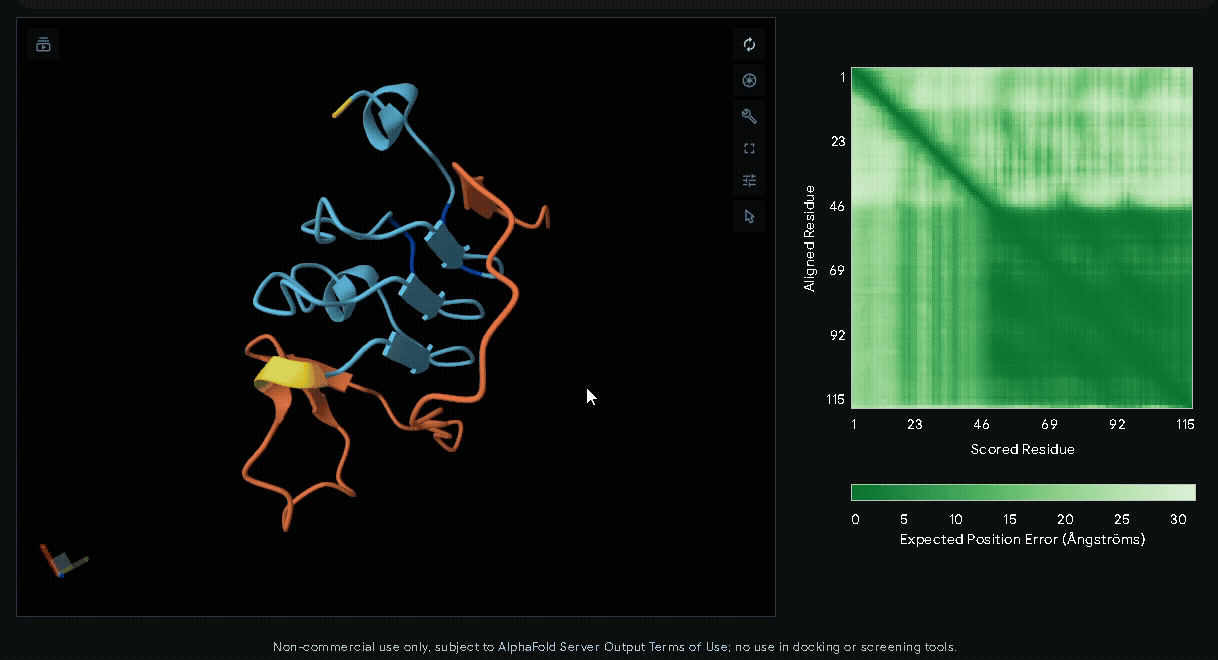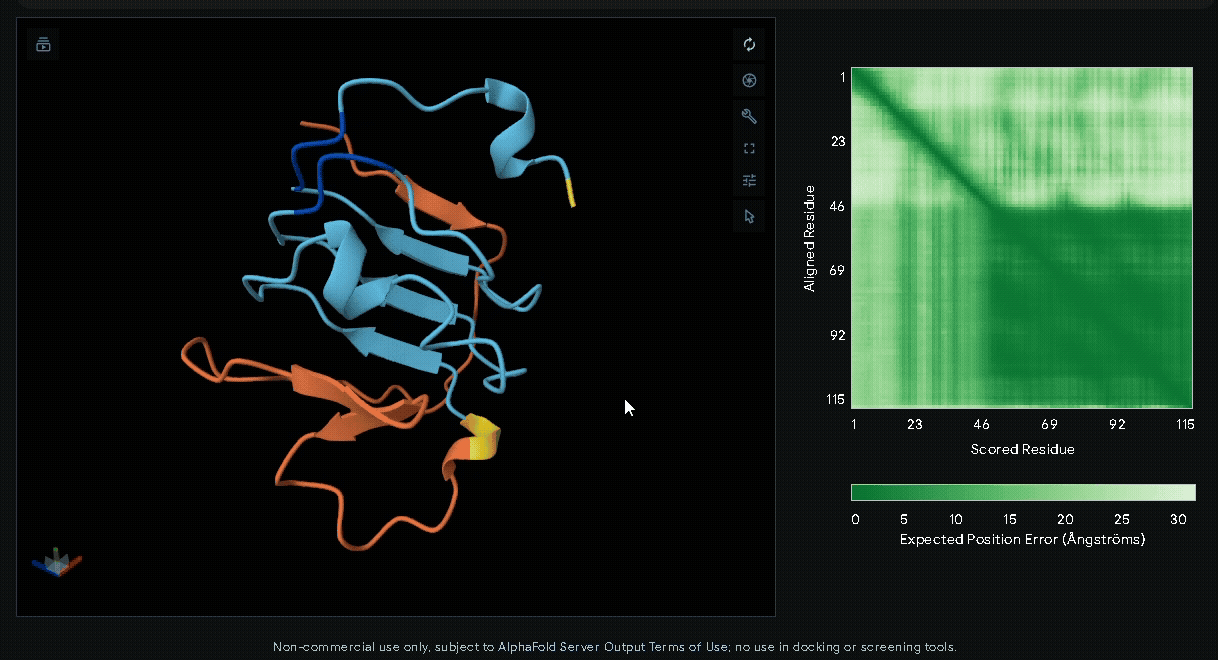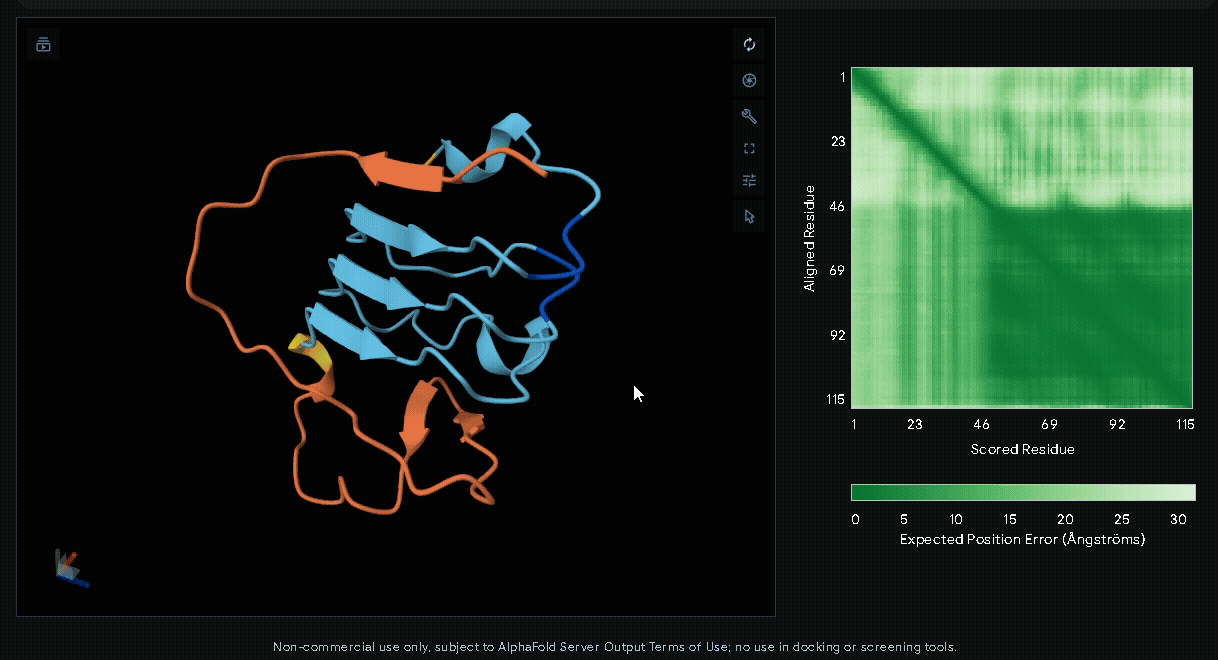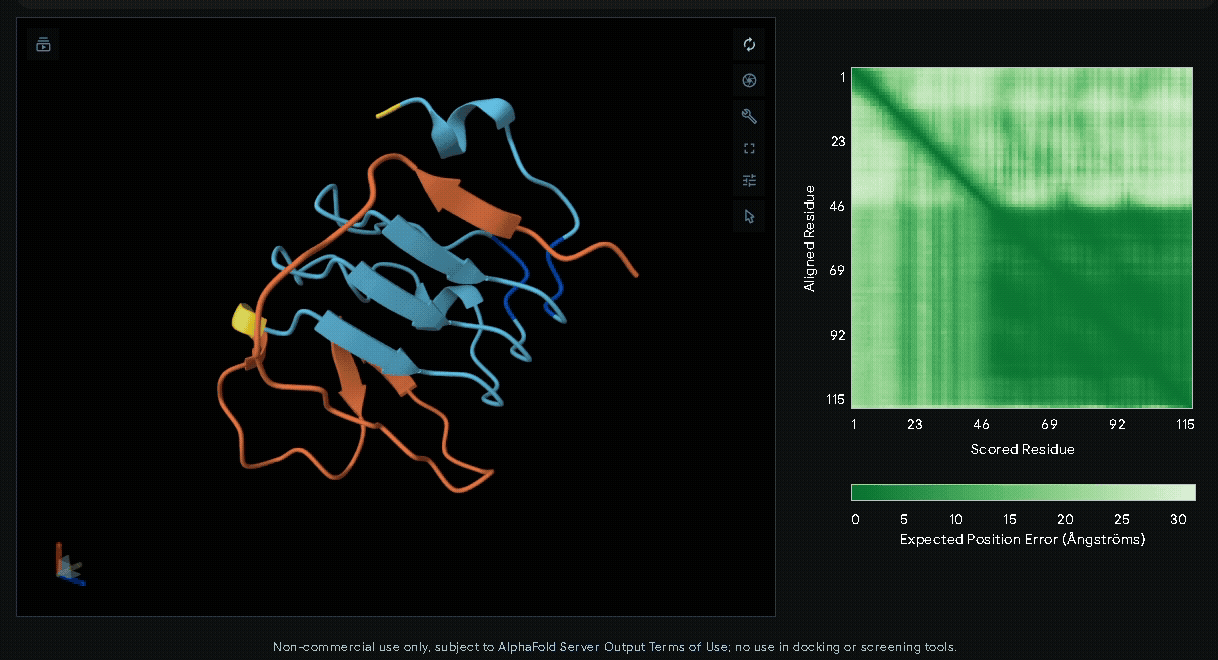
-
6xHis-(Nup-DnaEC)-CWE-CBD(Lumican)
A His-tag was introduced into the sequence to facilitate protein purification.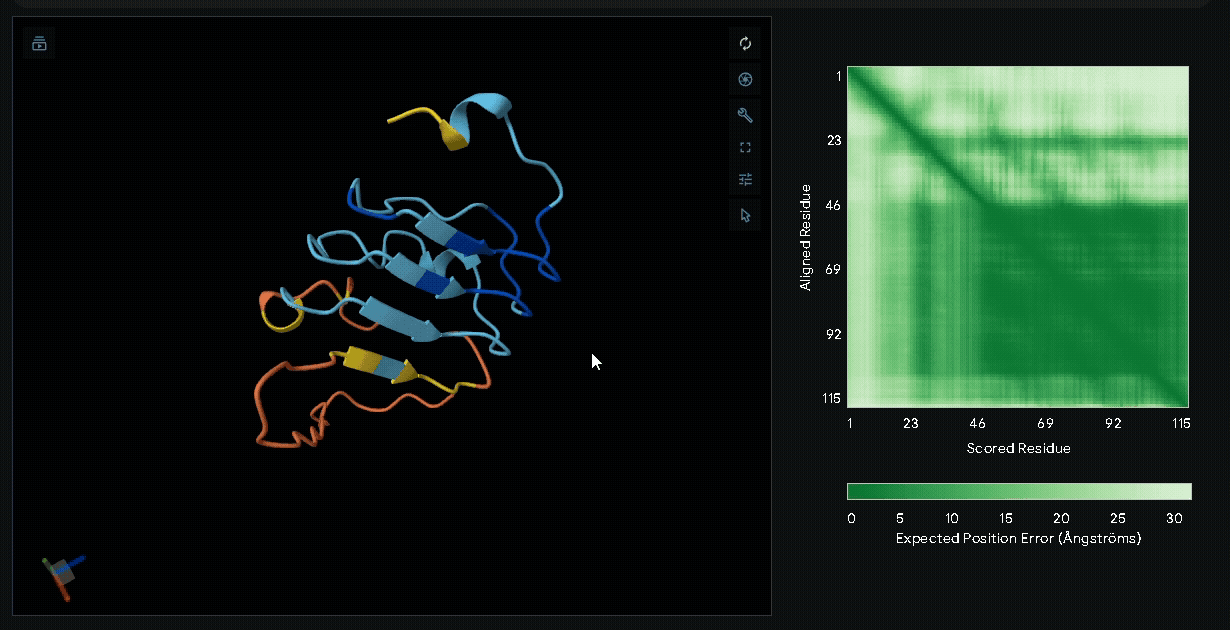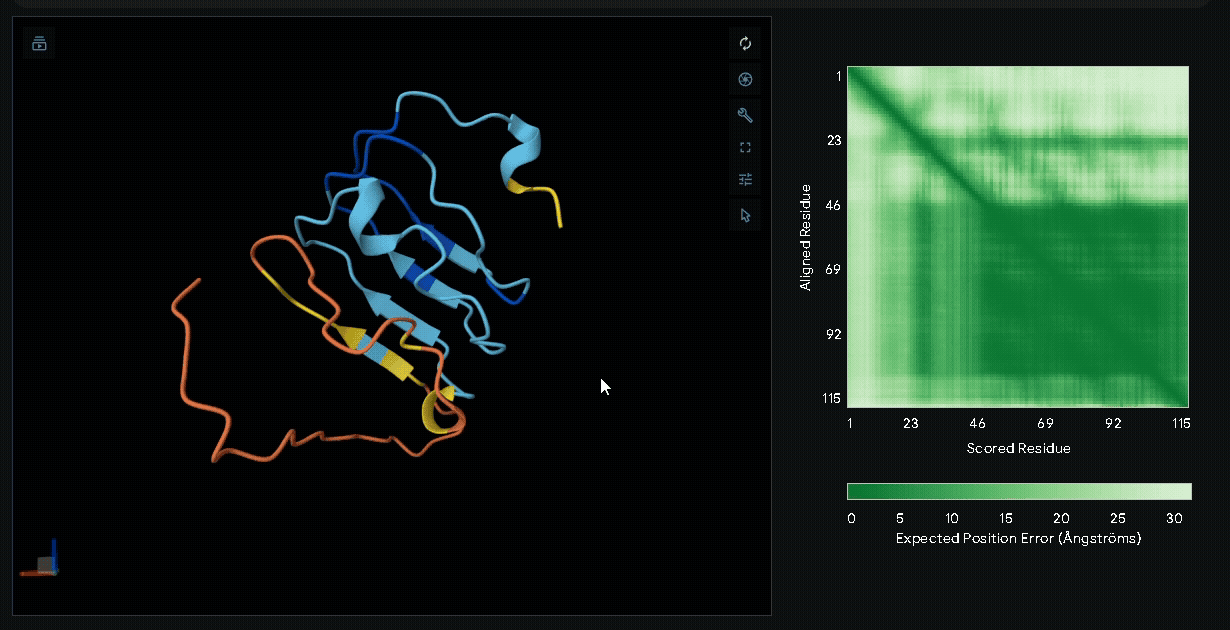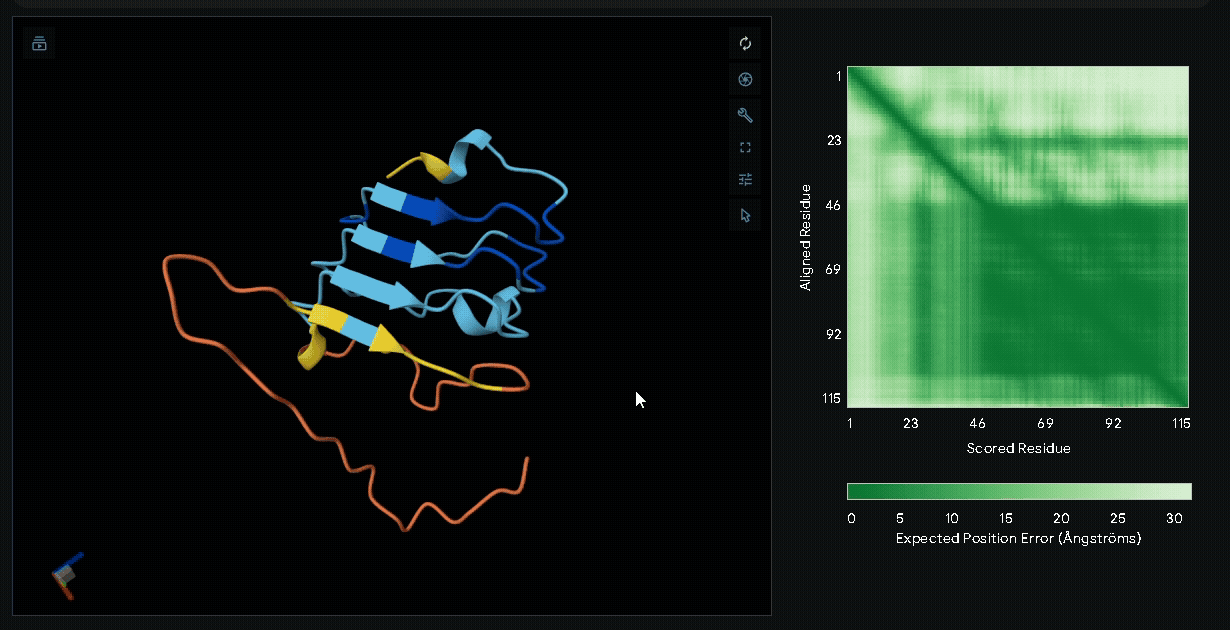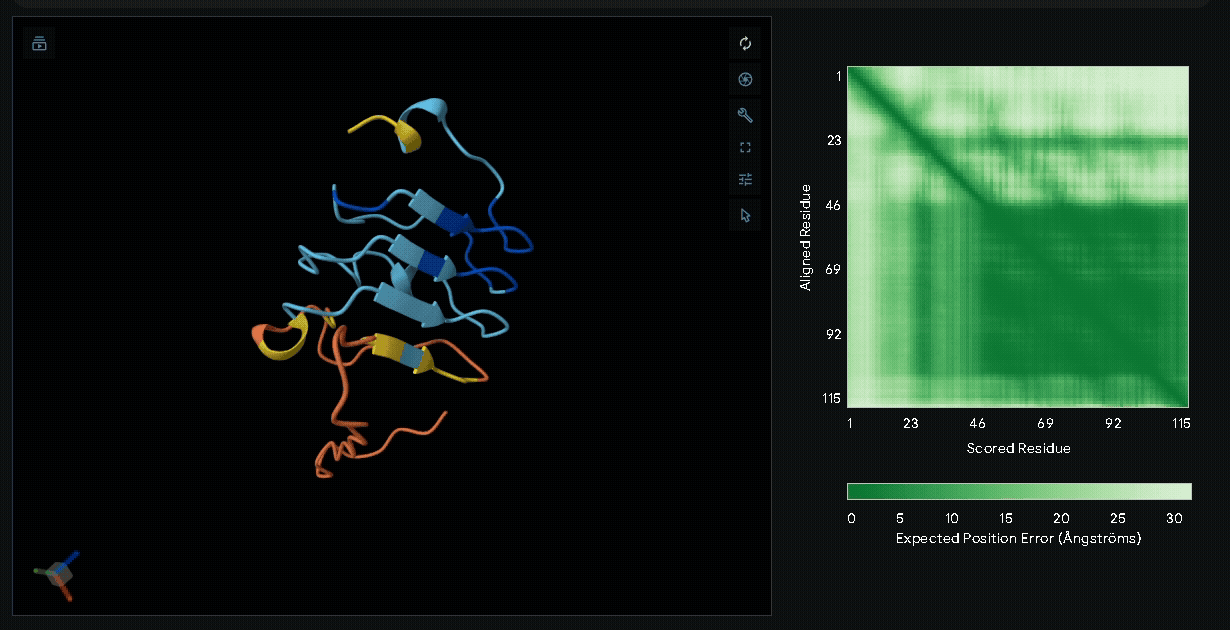
-
sRAGE-eGFP-6xHis-RGK-(Nup-DnaEN)
A His-tag was introduced into the sequence to facilitate protein purification, while EGFP was incorporated to verify successful transfection in mammalian cells and to visually confirm the surface attachment of our fusion proteins onto collagen hydrogels.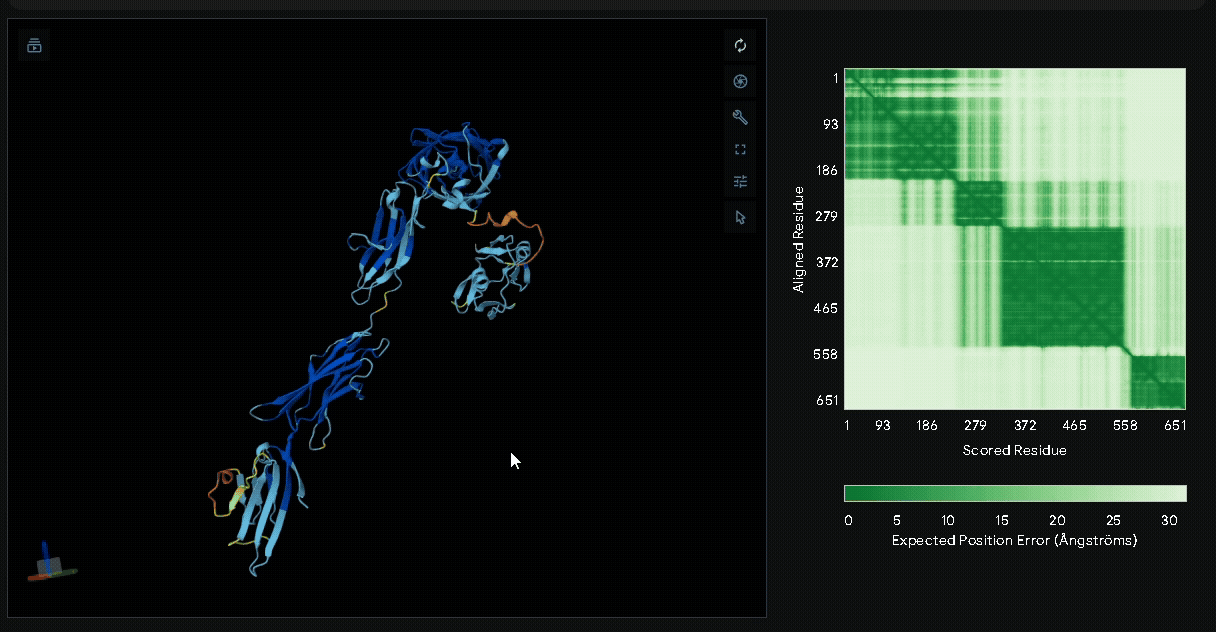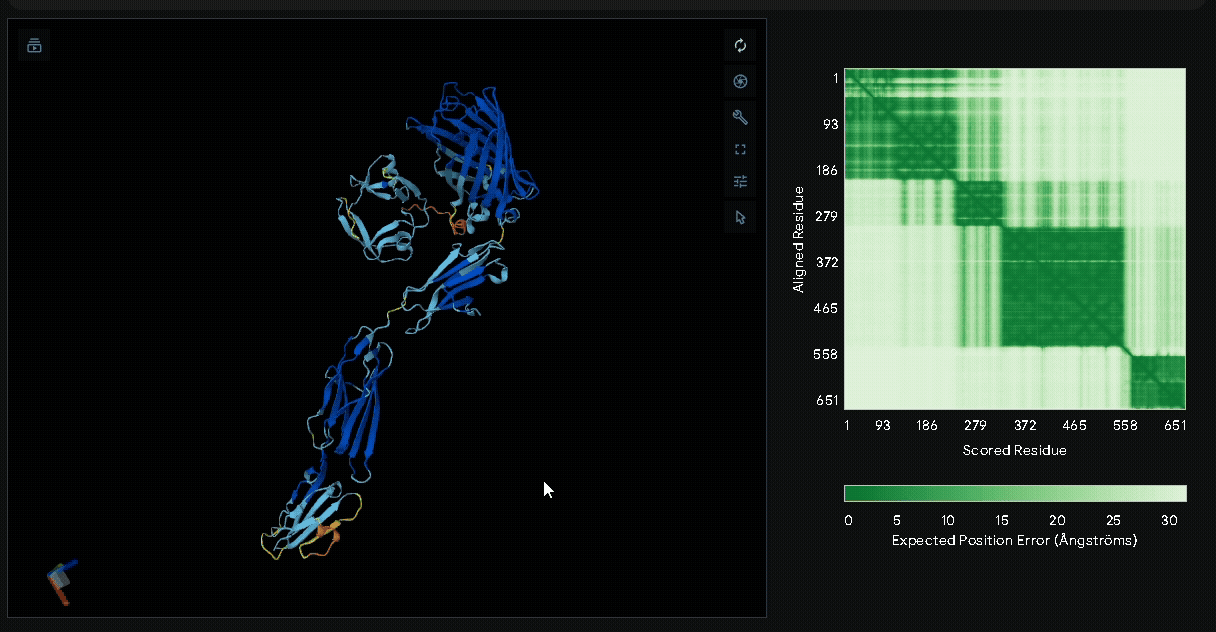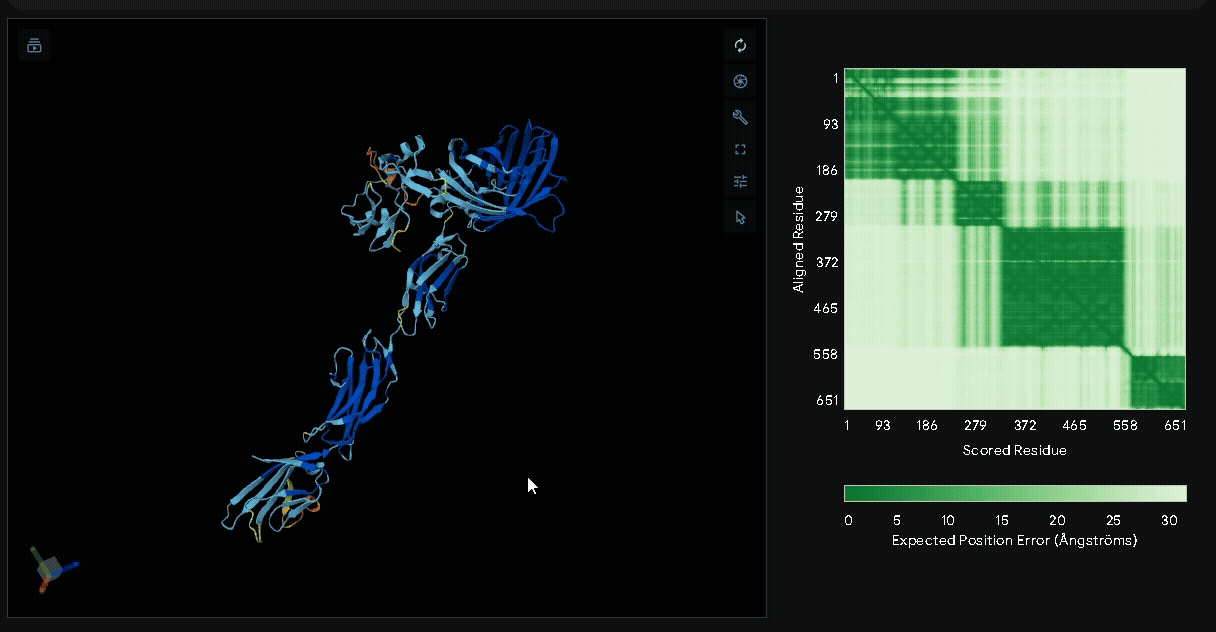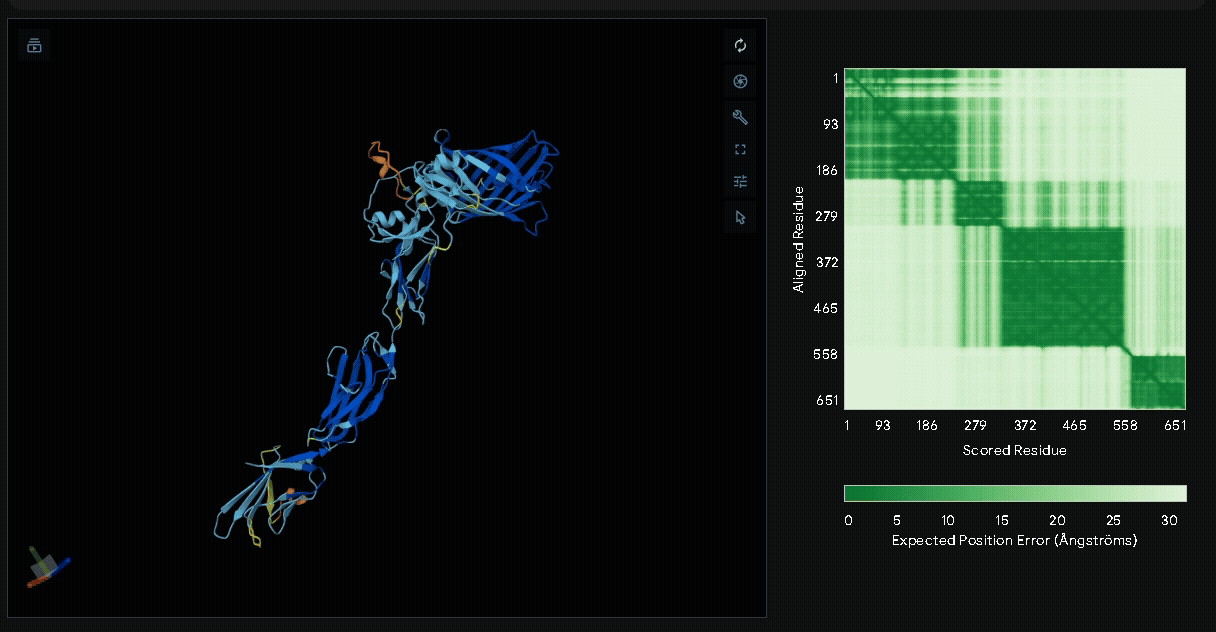
-
sRAGE-eGFP-6xHis-RGK-CWE-6xHis-CBD
Structural modeling of intein-spliced fusion proteins.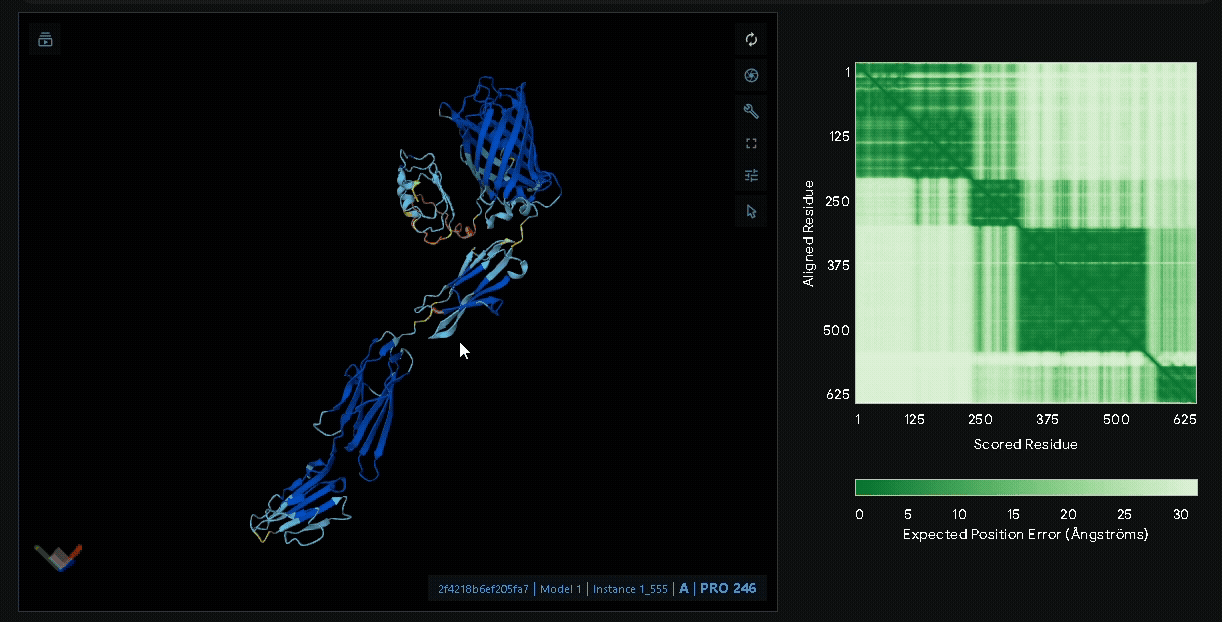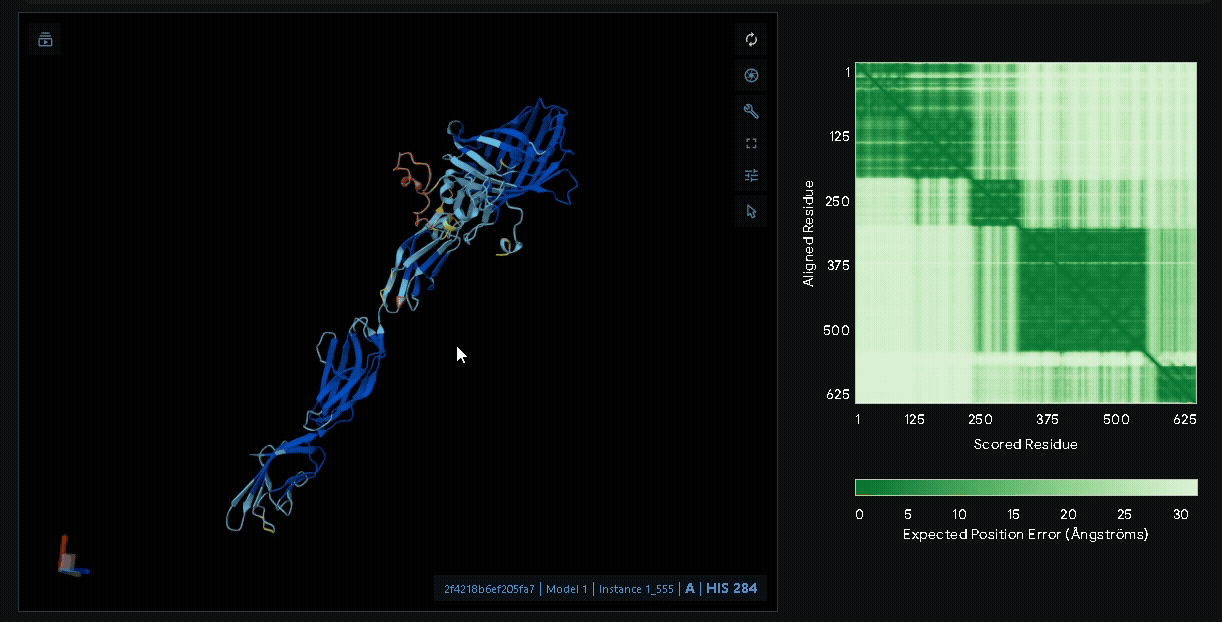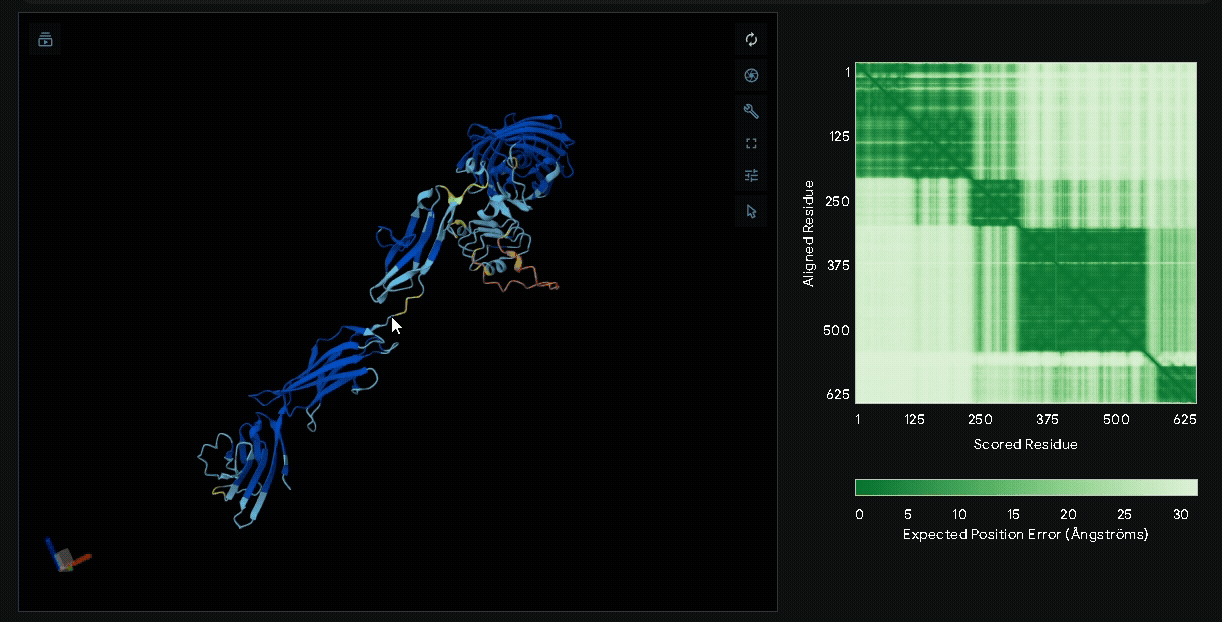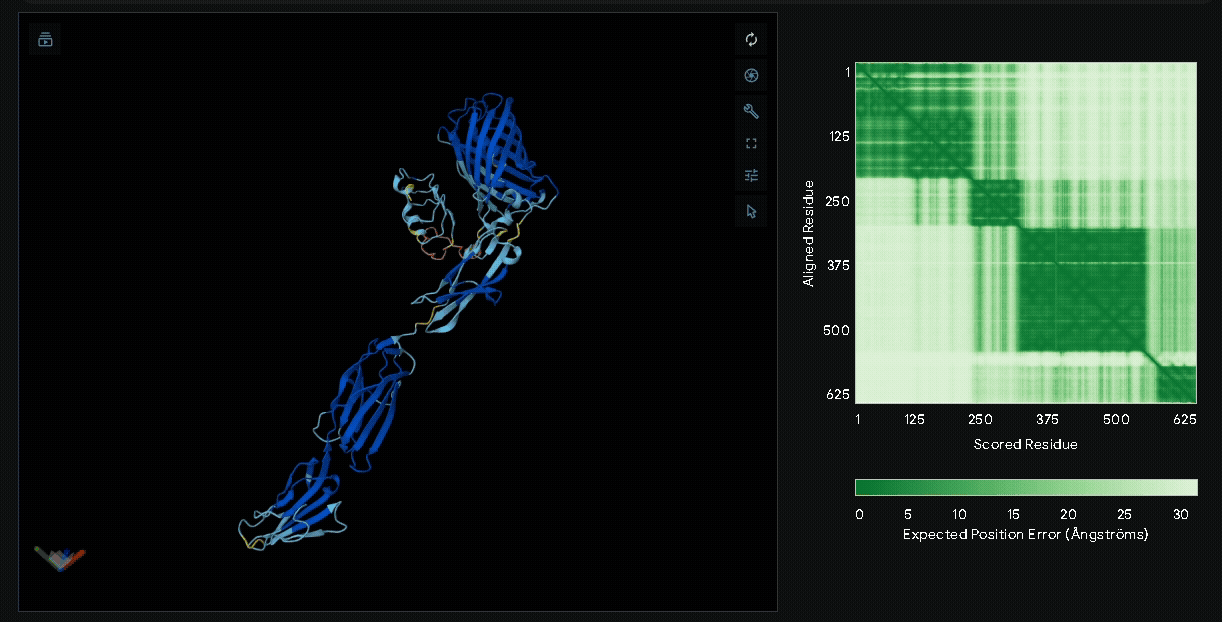
Conclusion
Through structural modeling with AlphaFold3, we build the structure of sRAGE and
its fusion proteins, particularly the lumican-based, no-linker design. These
predicted structures suggested proper folding and minimal inter-domain
interference. By integrating the Npu DnaE split intein system into our
protein structure modeling, we believed that a modular and switchable
platform of therapeutic proteins on is possible.
Since there are many types of AGEs with distinct molecular structures, future
work could involve modeling the interactions between various AGEs and our
fusion proteins, further optimizing binding specificity and therapeutic performance.
Reference
- Arai, R., Ueda, H., Kitayama, A., Kamiya, N., & Nagamune, T. (2001). Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein engineering, 14(8), 529-532.
- Bhandari, D. G., Levine, B. A., Trayer, I. P., & Yeadon, M. E. (1986). 1H‐NMR study of mobility and conformational constraints within the proline‐rich N‐terminal of the LC1 alkali light chain of skeletal myosin: Correlation with similar segments in other protein systems. European journal of biochemistry, 160(2), 349-356.
- Kalamajski, S., & Oldberg, Å. (2009). Homologous sequence in lumican and fibromodulin leucine-rich repeat 5-7 competes for collagen binding. Journal of Biological Chemistry, 284(1), 534-539.
- Iwai, H., Züger, S., Jin, J., & Tam, P. H. (2006). Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS letters, 580(7), 1853-1858.