Using LigandMPNN/ProteinMPNN coupled with energy minimization methods,
we enabled soluble expression of plant chitinase PrChiA in E.
coli with 2.407U/mg activity.
We demonstrated the application of Glide to dock large hexasaccharide
ligands to enzymes and developed a workflow for insilico screening of affinity of redesigned enzymes to
carbohydrates.
A total of eighteen variants for three enzymes were designed, evaluated
by an in silico matrix, and tested in wet lab.
Abstract
Central to our fungicide are two classes of glycoside
hydrolase, chitinase and glucanase, both of which act to sever and
disintegrate the fungal cell wall, leading to lysis of the cell. To improve
our product's feasibility and commercial value, we engineered three enzymes for improved
soluble yield, enzyme stability and activity by sequence
redesign.
We based the first round of dry lab engineering on
methods by Sumida et al., 2024
[1]. We modified the catalytic domains of the three
enzymes, PrChiA, GlxChiB and BglS27, using deep learning-based
tools after identifying evolutionarily conserved sites. Redesigned sequences
are then evaluated by a matrix containing their calculated protein structure
energies and / or ligand docking scores (Fig. 1).
Fig. 1 | Schematic for our model-guided design
process. Design tools take wildtype protein structure and selected
design space (residues to be redesigned) as input to generate novel protein
sequences that are then evaluated by a matrix. Best performing designs are then selected for wet lab
testing.
Our results demonstrate the potential of
deep-learning methods in enhancing protein solubility, with several designs
achieving soluble expression while their wildtype enzyme remains insoluble.
This is particularly useful for future iGEM teams facing issues of insoluble
expression of proteins in E. coli. During the course of our
engineering cycles, to more efficiently process the large scale of sequence
redesign and subsequent structure prediction and evaluation, we developed
several scripts for automating these tasks (see our
Supplementary material for detailed
recordings of our methodology and scripts). Our development of a carbohydrate
docking pipeline could also potentially assists future iGEM teams in their
modelling effort.
Together with our wet lab results, the
concerted development of our dry and wet lab effort paves road for better
understanding of the behavior of carbohydrate hydrolases as well as enhanced
commercial values of ArMOLDgeddon.
Introduction
In our project, we aim to produce glycoside
hydrolases, specifically chitinases and glucanases, for broad-spectrum
antifungal effects by targeting carbohydrates within the fungal cell wall.
Glycoside hydrolases typically have a modular architecture consisting of a
single catalytic domain connected to one or several carbohydrate binding
domains or modules (CBDs/CBMs) by flexible linker regions (Fig.
2a).
Fig. 2 | (a) General architecture of chitinase (left,
GlxChiB) and glucanase (right, BglS27) is composed of a globular catalytic
domain joined to one or more carbohydrate binding domains (CBM) by a
flexible linker; (b) Schematic of hydrolase hydrolyzing fungal cell wall
glycan, leading to lysis of the fungal cell.
Among the enzymes that we chose to characterize, some
showed unsatisfactory results. For chitinases, PrChiA were unable to
achieve soluble expression in E. coli, while GlxChiB showed little
hydrolysis activity towards colloidal chitin (Fig. 3a,c). For
glucanases, except ɑ-1,3-glucanase aglEK14, the three other glucanases
(FlGlu30, BglS27, and bglu1) showed good soluble expression yields
(Fig. 3b). However, BglS27 and FlGlu30 showed low
hydrolytic activity. Considering the role of β-1,3-glucan as a major structural component of fungal cell
wall, we decided to focus our dry lab effort on BglS27 for glucanases.
We therefore attempted model-guided redesign of catalytic
domains of three enzymes: PrChiA, GlxChiB, and BglS27 (Fig.
3d) to enhance their solubility, stability and activity.
Fig. 3 | (a, b) SDS-PAGE of wild type BcChiA1,
rMvEChi, GlxChiB, PrChiA, Bglu1, BglS27 and FlGlu30
with BL21(DE3) with empty vector as control (LB: supernatant after overnight
fermentation; wc: whole cell samples; s: supernatant after ultrasonic cell
lysis; p: pellet after ultrasonic cell lysis); (c) hydrolytic activity of
cell lysate supernatant of wildtype chitinases towards colloidal chitin.
PrChiA*: activity of PrChiA wildtype is not tested due to
inclusion body expression; (d) Enzyme specific activity of cell lysate
supernatant of Bglu1, BglS27, and FlGlu30, each with the bracketed
substance as substrate. Error bars represent±SD (n=3).
Fig. 4 | General workflow of our modelling effort.
Design space is selected after combining rational docking results with
evolutionary conservativeness (from multiple sequence alignment methods). Sequences
redesigned with LigandMPNN or ProteinMPNN are passed to AlphaFold3 to
generate respective variant structures, which are evaluated based on SASA,
Rosetta energy score, and Cα RMSD relative to wildtype structure. Sequences
performing best in silico are selected for wet lab testing. One
enzyme unit (U) is defined as the amount of enzyme required to to release 1
μmol of reducing sugar per minute at 37°C. s: cell lysate supernatant; p:
cell lysate precipitate; wc: whole cell; LB: culture liquid.
We derived the design pipeline from method by Sumida
et al., 2024
[1], which involves identifying enzyme active site
residues and evolutionarily conserved residues from iterative rounds of
multiple sequence alignment (MSA). Given those residues are predicted to be
important for enzyme stability and activity, they were fixed, and other
residues were redesigned by deep-learning protein design algorithm
ProteinMPNN to generate more stable variants. This method enabled them to
enhance TEV protease yield by an average of 20-fold and activity by up to
26-fold.
Our protein design pipeline similarly started off with
generating an MSA, but we incorporated an additional step of rational
docking of substrate to the enzyme structure. Our sequence redesign process
also made use of LigandMPNN, the ligand-aware version of ProteinMPNN. The
designs were then evaluated using a matrix combining Rosetta score, Cɑ RMSD,
docking scores, SASA (solvent accessible surface area), to select the best
in silico performing designs (Fig. 4)
[2][3][4][5][6].
Details about the sequence redesign tools
ProteinMPNN
[7] is a deep learning-based sequence redesign tool
renowned for its ability to generate sequences that fold into a
backbone geometry similar to the input structure, but with
enhanced stability and solubility. ProteinMPNN is used in our project
in hopes for enhanced protein stability and
solubility.
LigandMPNN
[8] is the ligand-aware version of ProteinMPNN that
has the ability to design sequences for protein binders of the input
ligand that will complex with the ligand in the same way as the input
protein-ligand complex. LigandMPNN is used in our project in hopes for
higher affinity of the enzymes toward their substrates and hence
enhanced catalytic activity.
Chitinase
Structure Acquisition and Rational
Docking
The two chitinases selected for catalytic domain redesign, GlxChiB
[9] and PrChiA
[10], have their apo-enzyme crystal structures solved
and
deposited in PDB databases (PDB: 7V91, 4RL3 respectively). To predict their
active sites and their interactions with chitin ligands, we acquired their
solved structures and structurally aligned them with solved structures of
structurally similar enzymes bound with chitin ligands. GlxChiB is aligned
with a GH19 enzyme complexed with a molecule of GlcNAc₄ (PDB: 3WH1, RMSD =
1.026Å) and PrChiA is aligned with a GH18 enzyme crystal structure
complexed with a molecule of GlcNAc₈ (PDB: 1EIB, RMSD = 2.566Å)
(Fig. 5). The ligands are then superimposed onto GlxChiB
and PrChiA crystal structures as per our knowledge-based rational
docking strategy.
Fig. 5 | Crystal structures of GlxChiB and
PrChiA complexed with template structures with other hydrolase of
the same protein family complexed with GlcNAc multimer (3WH1 with GlxChiB,
1EIB with PrChiA).
The superposed ligand-complexed structures were then
refined with Schrodinger Prime "refine protein-ligand complex" function and
energy-minimized by Rosetta FastRelax to remove bad contacts
[2][3][4].
Active Site Identification
Both chitinases belong to reasonably well-studied
protein families, GH19 for GlxChiB and GH18 for PrChiA. Catalytic
residues of GlxChiB were identified by superposing its crystal structure
with the crystal structure of a GH19 papaya chitinase (PDB: 3CQL,
RMSD=0.592Å), in which two glutamate was characterized as a catalytic residue
[11]. Through structural comparison, we located the
catalytic residues of GlxChiB to be E67 and E89 (Fig. 6a). Catalytic
residues of PrChiA were identified based on the characterized
catalytic motif of its family (DXXDXDXE, D116, D119, D121, E123) in
CAZypedia documentation on the GH18 protein family (Fig.
6b)
[12]. This information is cross-verified by inspecting
ligand-complexed structure of PrChiA for glycosidic bonds near the
reactive residues.
Fig. 6 | Close-up view of catalytic residues (side
chain shown as dark green sticks with labels) of (a): GlxChiB and (b):
PrChiA (green cartoon) in complex with chitin ligand (light green
stick). 3CQL, superposed with GlxChiB, is also shown (light gray cartoon).
With identified reactive residues, the 0 position
(glycoside bond to be hydrolyzed) and glycoside residues occupying -1 and +1
subsites (glycoside residue at reducing end and non-reducing ends of 0
position, respectively) were identified. The active site residues to be fixed in sequence redesign was
therefore considered residues with atoms within 5Å of
the two glycoside residues occupying -1 and +1 subsites.
Conserved Site Identification
Conserved sites were identified following method by
Sumida et al., 2024
[1]. Four iterative rounds of multiple sequence alignment
(MSA) by HHblits
[13] were performed for GlxChiB and PrChiA
querying their catalytic domain sequences against UniRef30 database with
increasingly inclusive E-value cutoffs at 1e-50, 1e-30, 1e-10, and 1e-4. The
final MSA after the fourth round of iterative search was then filtered for
90% sequence redundancy, 50% coverage, and 30% minimum identity. Each
individual position in the MSA was ranked by degree of conservation and the
top fifty-percent conserved positions were selected as conserved sites.
E-value
E-value is a measurement of how likely the
query and result are similar by chance instead of due to evolutionary
relationship. In general terms, it represents how similar the query
and the result are: the higher the similarity, the lower the E-value.
Lower E-value cutoffs can therefore be considered to lead to
"stricter" searches. In context of identifying conserved positions,
varying E-value cutoff between searches aims to produce a good representation of evolutionary
conservativeness of the query's protein family.
Sequence Redesign and Structure
Prediction
ProteinMPNN was used to redesign sequences for
variants with enhanced stability and solubility, Rosetta-FastRelaxed and
non-FastRelaxed chitinase structures without ligand atoms were used as
inputs. LigandMPNN was used to redesign sequences for variants with enhanced
catalysis. Similarly, Rosetta-FastRelaxed and non-FastRelaxed structures were used as inputs but including
the ligand atoms; only conserved site residues were fixed during the redesign.
Sixteen sequences were redesigned
at temperatures 0.1, 0.2, and 0.3 for each input structure for both ProteinMPNN and LigandMPNN,
generating 384 sequences.
Structures are then predicted using
AlphaFold3 at AlphaFold Server
[14].
AI and Physics-inspired methods
AI or deep learning-based methods for various
aspects of biomolecular modelling, such as structure prediction
(AlphaFold3, Chai-1, RFAA) and protein design (RFdiffusion,
ProteinMPNN, ESM3) arose in recent years and had quickly become
alternatives to traditional physics-inspired methods.
In
our project, we extensively employed these novel tools due to their
higher tolerance on accuracy of the structures used as their input and
also, in some cases, a lesser demand on computational resources and
expertise
[15].
Evaluation of Redesigned Variants
We evaluated redesigned chitinase variants by
modelling them with AlphaFold3 and scoring them based on solvent
accessible surface area (SASA, higher value suggests better solubility), Cα
RMSD (indicator of difference between backbone geometry between wildtype and
designed variant; lower is better), and Rosetta energy score (indicator of
stability of the protein; a more negative value is better)
[3][4][14]. Due to similarities of RMSD and SASA between the
variants, 12 variants (6 for each enzyme) with the best Rosetta energy scores were chosen for
wet lab testing (Table 1).
Table 1 | RMSD, ΔΔG, and SASA of representative
designs of PrChiA and GlxChiB
Name
Cα RMSD
ΔΔG /REU
SASA /Å2
ΔG /REU
Wet lab Label
PrChiA-Protein-t02-11
0.424
-39.5
11022
-657.9
PrChiA-3
PrChiA-Relaxed-Protein-t01-5
0.646
-30.6
10956
-649.0
PrChiA-5
PrChiA-Relaxed-Protein-t02-11
0.647
-26.8
10957
-645.2
PrChiA-6
PrChiA-Relaxed-Protein-t01-16
0.638
-24.1
11073
-642.6
N/A
PrChiA-Relaxed-Protein-t01-13
0.666
-23.4
11140
-641.9
N/A
PrChiA-Relaxed-Ligand-t02-4
0.623
-77.7
10855
-696.1
PrChiA-1
PrChiA-Relaxed-Ligand-t02-14
0.680
-62.1
10813
-680.5
PrChiA-2
PrChiA-Relaxed-Ligand-t02-5
0.670
-51.5
11130
-670.0
PrChiA-4
PrChiA-Relaxed-Ligand-t01-10
0.575
-47.6
11226
-666.1
N/A
PrChiA-Relaxed-Ligand-t03-9
0.667
-44.9
10929
-663.3
N/A
GlxChiB-Protein-t01-6
0.391
-105.3
10756
-422.4
GlxChiB-3
GlxChiB-Relaxed-Protein-t03-10
0.720
-104.0
10795
-421.1
GlxChiB-6
GlxChiB-Protein-t02-5
0.421
-102.7
11125
-419.8
GlxChiB-4
GlxChiB-Relaxed-Protein-t03-13
0.708
-97.7
10629
-414.8
N/A
GlxChiB-Relaxed-Protein-t02-10
0.492
-89.8
10620
-407.0
N/A
GlxChiB-Relaxed-Ligand-t03-3
0.498
-102.3
10576
-419.4
GlxChiB-1
GlxChiB-Relaxed-Ligand-t01-7
0.741
-99.0
10822
-416.1
GlxChiB-2
GlxChiB-Ligand-t01-16
0.521
-90.3
10692
-407.4
GlxChiB-5
GlxChiB-Ligand-t03-13
0.402
-88.8
10782
-405.9
N/A
GlxChiB-Relaxed-Ligand-t02-6
0.649
-83.7
10716
-400.8
N/A
For a single enzyme,
Candidates with
name [Enzyme]-Protein were generated with ProteinMPNN, while [Enzyme]-Ligand
were generated with LigandMPNN. Temperature and identifier in each
temperature-batch of the designs are also appended to the names (t0x-y denotes yth redsigned
sequence of temperature 0.x). Rows shaded in green were chosen for
testing in wet lab experiments. ΔG and ΔΔG are the Rosetta energy score
computed for the variant and difference between ΔG with the wildtype's
Rosetta energy score. Wet lab labels are the shorter label used in wet lab
experiments for simplicity; solubility will be reported using these labels.
REU=Rosetta energy unit, arbitrary energy unit of Rosetta.
Results
Due to time constraints of our iGEM project, six best
ranked designs for each of PrChiA and GlxChiB were tested in wet lab through
expression in E. coli BL21(DE3), with the original catalytic domain
replaced by redesigned variants (Fig. 7).
Fig. 7 | Expression cassette of PrChiA and
GlxChiB; the catalytic domains are joined to the wildtype N-terminal set of
CBMs and linkers and to a 6×His tag at the C terminal.
Compared to wildtype PrChiA displaying only
insoluble expression, five of the six designed PrChiA variants
(variants 1, 2, 3, 5, 6) achieved soluble expression. PrChiA-6,
with highest soluble expression yield of 0.262 g/L in supernatant of cell
lysate, shows a yield higher than all wildtype chitinases except from
rMvEChi (Fig. 8).
Fig. 8 | (a) SDS-PAGE of GlxChiB, and PrChiA,
with supernatant of BL21(DE3) with empty vector as control; (b) SDS-PAGE of the
six variants of PrChiA, with supernatant of BL21(DE3) with empty vector
as control; (c) soluble expression yield of each of the PrChiA
variants and wildtypes of BcChiA1, rMvEChi, GlxChiB, and
PrChiA. s: cell lysate supernatant p: cell lysate precipitate; wc:
whole cell; LB: culture liquid. Error bars represent±SD (n=3).
Chitinolytic activity of cell lysate supernatant is
assayed with a colorimetric method using colloidal chitin as substrate. 1 μL of cell lysate is added
to 200 μL of 2 mg/mL and incubated at 37°C for 10 min. Reducing sugar
remaining in the reaction mixture is quantified by measuring increase in
absorbance at 540 nm after reaction with 3,5-Dinitrosalicylic acid (DNS)
(Miller, 1959). The experiment is done in triplicate.
[16].
In the assay, cell lysate of two
PrChiA variants shows catalytic activity against colloidal chitin,
whereas the wildtype does not display any due to inclusion body expression.
The best performing variant is PrChiA-3 with 2.407 U/mg activity
(Fig. 9). One enzyme unit of activity (U), is defined as
the amount of enzyme required to release 1 μmol of reducing sugar per minute
from colloidal chitin under 37°C.
Fig. 9 | Chitinolytic of cell lysate supernatant of
the six designed variants of PrChiA with colloidal chitin as
substrate; PrChiA-4*: chitinolytic activity of PrChiA-4 is
not measured due to inclusion body expression of the variant. Error bars
represent±SD (n=3).
Results for GlxChiB are less satisfactory. While all
six variants are successfully expressed in BL21(DE3), none had achieved
soluble expression, perhaps due to the enzyme's eukaryotic origins
(Ficus macrocarpa, an evergreen tree).
Glucanase
Design method for glucanase was similar to our method
for chitinase design, with an extended scoring matrix including docking
scores of AlphaFold3-predicted structure of the redesigned variants and a modified design
space selection method (Fig. 10).
Fig. 10 | Flowchart displaying design process of
glucanase. Design space is similarly determined using MSA and docking
results, however modified setups of design space selection
(redesigning entire active site or binding site) are included in addition to
those employed during the chitinase design process. Designs from LigandMPNN
and ProteinMPNN are passed to AlphaFold to generate structure of the
designs. Redocking of the ligands back to structures of the designed
variants yield an extra docking score term that is used along with SASA,
Rosetta energy score, and Cα RMSD to evaluate performance of the designs.
The best in silico -performing designs are selected for wet lab testing.
Structure acquisition
Since there is no solved crystal structure, BglS27
structure was modelled with AlphaFold3 and β-1,3-glucan hexamer was docked
to its active site using Glide. Docking grid is constructed based on
position of glucan in 1U0A (A catalytically inactive GH16 glucanase mutant
complexed with a beta-glucan tetrasaccharide). The docked enzyme-ligand
complex is then refined with Schrodinger Prime "refine protein-ligand
complex" function (refinement limited to within 5Å of ligand) followed by
Rosetta FastRelax. The final structure from FastRelax is used as input for
sequence design tools.
Docking carbohydrates
We carried out a simple benchmarking of the
three docking engines, Vina
[17], Vina-Carb
[18], and Glide
[6] by docking substrate glucans into the
AlphaFold-predicted wildtype structure of three glucanase used in our
project. (BglS27 (β-1,3-glucan tetramer), Bglu1 (β-1,3-glucan
tetramer), and FlGlu30 (β-1,3-glucan tetramer, with β-1,6
linkage instead of 1,3 linkage in between the two central glucose; not
shown in diagram)) Results suggests that Vina and Vina-Carb seem to be
unable to place the ligand across the entire ligand-binding cleft
(Fig. 11), even at high exhaustiveness (a parameter
of the two docking engines that determines the amount of computational
power used for docking; higher exhaustiveness means more computational
power; docking was ran with exhaustiveness set to 160 and with 8
threads allocated to a single docking run it takes ~30 min to complete
for both Vina and Vina-carb to complete a structure).
On
the other hand, while at default settings Glide is unable to report
any docked pose (i.e. all rejected by the engine), it is found that
increasing the "poses kept per ligand for initial phases of docking"
parameter to 800000 and enabling "use extended sampling" along with
using XP mode managed to produce poses that can occupy most of the
binding cleft while spending only around 1.5 hours on a single thread
to complete a structure.
We therefore chose Glide for both
docking glucan to BglS27 to produce the initial protein-ligand complex
used for LigandMPNN input and for the later redocking process due to
its high accuracy and high throughput.
Fig. 11 | Docking results of β-1,3-glucan
tetramer to Bglu1 and BglS27 wildtype enzymes from different docking
engines. Only the most negative scoring (best deemed by the engines)
pose for each engine is shown. Docking pose from Vina-Carb, Vina, and
Glide are shown in dark green, green, and light green, respectively.
Design space selection
Evolutionarily conserved sites were similarly
identified using HHblits, however, it is noticed top 50% conserved sites
encompass the entire active site of the enzyme and almost the entire
ligand-binding cleft (Fig. 12a). In an attempt to
experiment with a different design method, we arrived at three different
design space selection strategies: fixing non-active site and catalytic
residues, fixing non-binding site and catalytic residues, and fixing top 50%
conserved residues. (Table 2) Catalytic residues were
identified based on the GH16 subgroup 1 catalytic motif (EXDXXE, residues E124 to
E129 in BglS27) suggested by CAZypedia
[19] (Fig. 12).
Fig. 12 | (a) BglS27 complexed with docked
β-1,3-glucan hexamer. Side chain of catalytic residues is shown in dark
green and top 50% conserved residues are colored green; (b) Close-up view of
catalytic residues (side chain shown as dark green sticks with labels) of
BglS27; the enzyme is in complex with β-1,3-glucan hexamer.
Table 2 | Three design space
selection methods, their
fixed residue selection criteria, and the aim of each method
Label
MPNN Model
Redesigned Residues
Aim
Protein
ProteinMPNN
Non-active site and non-top 50% conserved residues
Enhance stability of the enzyme through redesign of non-essential
residues
Ligand-AS
LigandMPNN
Active site (AS, residues within 5Å of -1 and +1 glucose) residues
except catalytic residues
Enhance enzymatic activity through redesigning the active site
Ligand-BS
LigandMPNN
Binding site (BS, residues within 5Å of the β-1,3-glucan hexamer)
residues except catalytic residues
Enhance enzymatic activity through redesigning the entire ligand
binding site
Sequence redesign
Sequence redesign is similarly carried out using
ProteinMPNN and LigandMPNN. LigandMPNN is used to redesign sequences for
design space selections fixing non-active site/non-binding site and
catalytic residues, at temperatures 0.1, 0.2, and 0.3, generating 20
sequences per temperature. ProteinMPNN is used to redesign sequences for
design space selection fixing top 50% conserved residues, again at three
temperatures, 20 sequences per temperature.
Evaluation of designs
All redesigned sequences from Ligand and ProteinMPNN
were passed to AlphaFold3 to generate structures for each
variant.
The predicted structures were all evaluated with the same
matrix used for chitinases: encompassing SASA, Cα RMSD relative to wildtype
structure, and ΔΔG relative to wildtype structure. In addition, docking of
β-1,3-glucan hexamer back to the variant structures (Fig.
13) is also included for designs generated by LigandMPNN, and the
docking scores are included in the matrix.
Fig. 13 | Redocking of β-1,3-glucan hexamer is carried
out in steps 1: superposition of wildtype docked protein-ligand complex
(template structure) with structure being evaluated 2: construction of a
grid (inner box 10Å, 46Å) with grid center at centroid of ligand atoms in
the template structure 3: Ligand is docked to the generated grid and
reported docked protein-ligand structure is manually inspected before
reporting the docking score to the final matrix
ProteinMPNN redesign of BglS27-Ligand-BS-t03-15
LigandMPNN-redesigned BglS27 variants show
unsatisfactorily positive ΔG values. Therefore, a second
round of ProteinMPNN redesign is applied to the BglS27 design with best
docking score, BglS27-LigandBS-t03-15 (LigandBS=design space encompassing
the entire binding site, see Table 2). AlphaFold-predicted structure of
BglS27-LigandBS-t03-15 is used as input for ProteinMPNN, fixing binding site
and top 50% conserved residues.
Predicted structures are again
evaluated with SASA, Cα RMSD relative to the wildtype, ΔΔG relative to
the wildtype, and docking score from redocking of β-1,3-glucan back to the
structure.
Finally, a total of six BglS27 designs from both first
and second round of design are selected to proceed to wet lab testing
(Table 3).
Table 3 | Docking score, RMSD,
ΔΔG, and SASA of
representative designs of the BglS27 variants
Name
Docking score /kcalmol-1
Cα RMSD /Å
ΔΔG /REU
SASA /Å2
ΔG /REU
Wet lab Label
BglS27-Protein-t01-4
N/A
0.533
-37.5
10251
-457.6
BglS27-1
BglS27-Protein-t02-18
N/A
0.543
-36.0
10134
–456.1
BglS27-2
BglS27-Protein-t02-8
N/A
0.520
-34.4
10389
-454.5
BglS27-3
BglS27-Protein-t02-15
N/A
0.520
-34.2
10156
-454.3
BglS27-4
BglS27-Protein-t03-10
N/A
0.518
-32.7
10493
-452.8
N/A
BglS27-Protein-t01-3
N/A
0.564
-32.7
10138
-452.7
N/A
BglS27-Protein-t01-7
N/A
0.530
-32.1
10267
-452.2
N/A
BglS27-BS0315-Protein-t03-1
-17.1
0.629
+18.1
10193
-402.0
BglS27-5
BglS27-Ligand-BS-t03-15
-19.8
0.546
+35.7
10218
-384.4
BglS27-6
BglS27-Ligand-AS-t02-6
-19.5
0.505
+78.5
10140
-341.6
N/A
BglS27-Ligand-AS-t02-1
-19.3
0.529
+60.6
10125
-359.5
N/A
BglS27-BS0315-Protein-t02-12
-16.6
0.636
+70.4
10102
-374.1
N/A
Variants with name
BglS27-Protein were generated with ProteinMPNN, while BglS27-Ligand were
generated with LigandMPNN. BS denotes design space encompassing the entire
binding site while AS denotes design space containing only the active site.
BS0315 indicates the design to come from the second round of ProteinMPNN
sequence redesign with BglS27-Ligand-BS-t03-15 as input structure. ΔG and
ΔΔG are the Rosetta energy score computed for the apo-enzyme structure of the
variant, and the difference between ΔG of the variant and that of the wild
type apo-enzyme structure. Temperature and identifier in each
temperature-batch of the designs are also appended to the names. Rows shaded
in green were chosen for testing in wet lab experiments. REU=Rosetta energy
unit, arbitrary energy unit of Rosetta. Wet lab labels are the shorter
labels used in wet lab experiments for simplicity.
Results
All six BglS27 designs were expressed in BL21(DE3)
and protein expression is assessed by SDS-PAGE of different cell
fractions (Fig. 14).
Fig. 14 | (a) Redesigned BglS27 catalytic domain is
expressed in E. coli with a C terminal His tag; (b) SDS-PAGE of six variants
of BglS27 with BL21(DE3) with empty vector as control.
Although redesigned BglS27 variants were successfully
expressed, none of them achieved soluble expression. Due to technical
limitations, inclusion body purification and refolding was not attempted and
hence activity of the designs is unfortunately not tested.
ProteinMPNN Redesign of BglS27 and
GlxChiB
Since the redesigned BglS27 and GlxChiB variants were
unable to achieve soluble expression, a second round of redesign is
performed on the wild type proteins with SolubleMPNN, ProteinMPNN trained on
a data set with only soluble proteins
[20]. 20 sequences are generated at each of the three
temperatures as before, and AlphaFold3 is used to predict structure of the
designs.
Fig. 15 | Scatter plot showing distribution of change
in Rosetta energy score relative to wildtype (X-axis) and change in SASA
relative to wildtype (Y-axis) of the wet lab designs and new SolubleMPNN
designs.
Due to time constraints, none of the redesigned
variants were tested in wet lab, but several BglS27 designs show greatly
enhanced SASA and ΔΔG values compared to the BglS27 designs tested
in wet lab, suggesting their potential to achieve soluble expression
(Fig. 15).
Discussion
Wet lab results of PrChiA
and GlxChiB
Our wet lab results show greatly enhanced protein
solubility for five out of six redesigned PrChiA variants expressed
in heterologous host E. coli, whose wildtype previously achieves only
inclusion body expression. Two of the redesigned variants achieved
functional expression and possess hydrolytic activity towards colloidal
chitin.
Compared to reported chitinolytic activity by
[21], the best-performing redesigned variant of
PrChiA, PrChiA-3, shows a two-magnitude lower activity
compared to the wildtype enzyme. However, this can be explained considering
that purified enzymes and different substrates (colloidal chitin in our
assay and soluble glycol chitin in literature) are used in the literature.
Brief research on comparison of hydrolytic activity of chitinase against the
two types of chitin substrate suggests that under the same conditions, at
least for GH18 chitinase proteins, glycol chitin is more readily
hydrolysable than colloidal chitin due to the former's higher solubility
[22].
For GlxChiB, although all six variants
are successfully expressed in BL21(DE3), none had achieved soluble
expression, perhaps due to the enzyme's eukaryotic origins. (Ficus
macrocarpa, an evergreen tree) Future experimentation with different
E. coli strains and different fermentation protocols might allow
soluble expression of the enzyme.
Interestingly, the designed
PrChiA variant with highest chitinolytic activity,
PrChiA-3, is designed by ProteinMPNN rather than LigandMPNN. This
is likely due to its enhanced protein stability and folding fidelity, which
will lead to higher enzyme yield in supernatant. A comparison between
Rosetta-calculated ΔG for designed variant structures and soluble expression
yield of soluble-expressed designed PrChiA variants suggests a
surprisingly positive correlation between calculated ΔG and expression yield
(R²=0.7726, Fig. 15B). Calculated SASA for the variants, however, are
positively correlated to soluble expression yield, as expected (Fig.
16a).
Fig. 16 | (a) Scatter plot of correlation between
calculated ΔG of PrChiA designs and their soluble expression yield;
(b) scatter plot of correlation between calculated SASA of PrChiA
designs and their soluble expression yield.
Design of carbohydrate hydrolase with ProteinMPNN and
LigandMPNN
We successfully achieved soluble and functional
expression of a heterologous plant chitinase, PrChiA, that previously
can only achieve inclusion body expression in E. coli, via a protein
design pipeline with ProteinMPNN, LigandMPNN, and energy-minimization
methods. We also demonstrated the potential of applying ProteinMPNN and
LigandMPNN to carbohydrate hydrolases and greatly enhancing the proteins'
soluble expression. Finally, we explored and demonstrated the usage of Glide
for docking extra-large hexasaccharide ligands to receptors and developed a
workflow for in silico screening of carbohydrate hydrolases using
the docking engine. Our wet lab results laid the foundation for further
improvements to the three enzymes, allowing methods such as directed
evolution and further sequence redesign with SolubleMPNN to further enhance
activity and solubility of the current designs.
Despite usage of
LigandMPNN, our dry lab effort on PrChiA did not yield large
improvements in enzyme efficacy. This might be explained by the general lack
of carbohydrate-interacting protein structures in the training of the LigandMPNN model.
We believe the results can also be due to a poor protein-ligand complex
structure that is used as input for LigandMPNN, as a result of purely in
silico methods.
However, it is evident ProteinMPNN and
LigandMPNN have the potential to greatly enhance solubility of enzymes, as
shown by the large increase in soluble expression in PrChiA designs
relative to the wildtype enzyme. Inclusion body expression for BglS27 is
likely due to importance of non-catalytic domains in solubility of the
enzyme being overlooked such that expressing the catalytic domains alone
will yield unsatisfactory results. Due to technical limitations of our lab,
we are unable to attempt renaturing and purification of the insoluble
GlxChiB and BglS27 designs which otherwise will allow evaluation of whether
the modified design space selection method employed for BglS27 will lead to
better results.
Reviewing calculated SASA and Rosetta energy
scores (Fig. 17) of the redesigned variants selected for
wet lab, it is noted that PrChiA variants possess especially large
increases in SASA, which likely explains their enhanced soluble expression
yield. This validates our in silico screening criteria and laid the
methodological foundation for future redesign efforts.
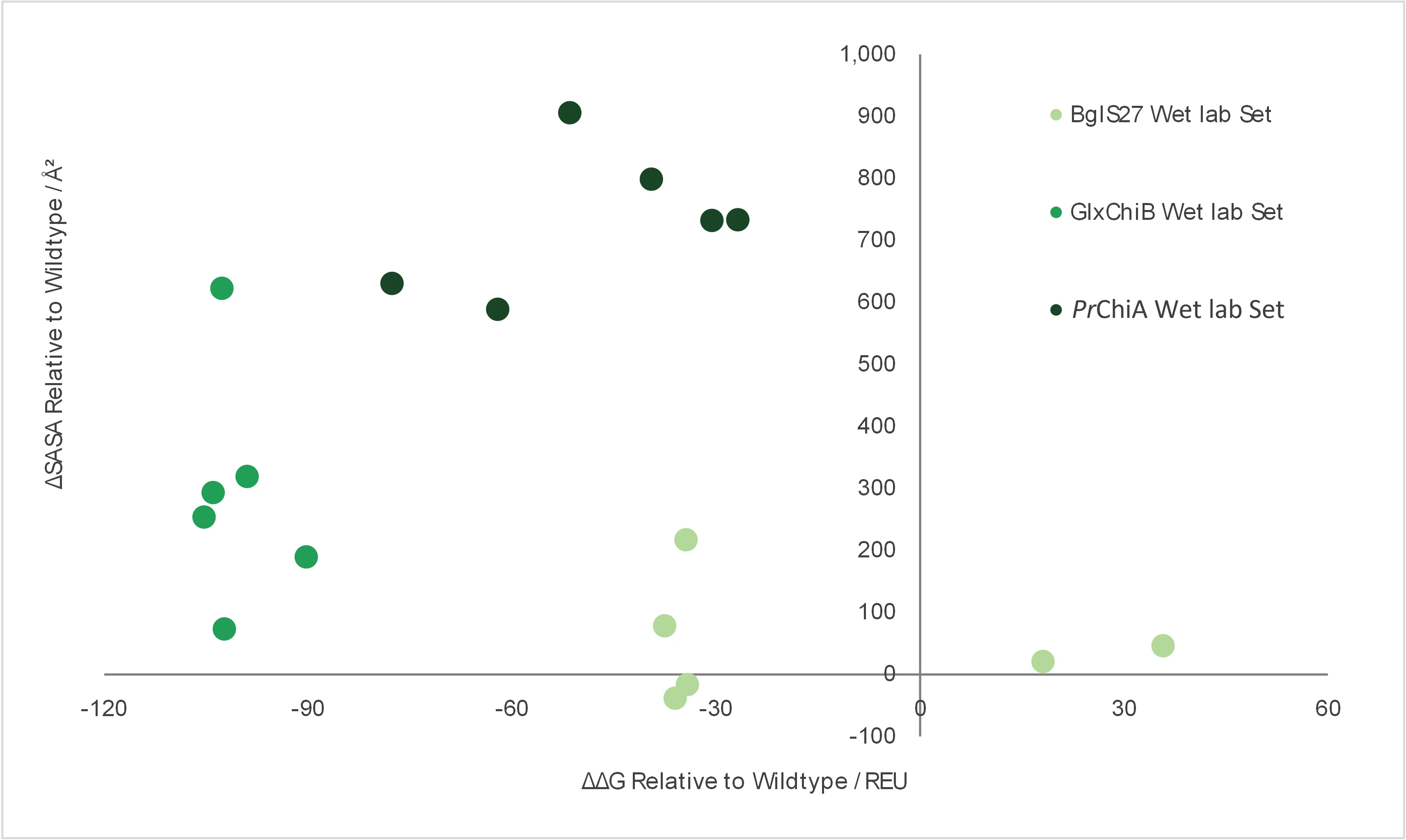
Fig. 17 | comparison of dry lab parameters (ΔSASA and
ΔΔG) of the enzyme candidates chosen for wet lab validation.
Contrasting to TEV protease
[1] and non-heme iron enzyme
[23] that are previous successful cases of ProteinMPNN
and LigandMPNN redesign, our multi-domain glycoside hydrolases are more complex and perhaps more caution
should be taken towards truncation of
non-catalytic domains. We also reflected that protein-ligand
complexes derived from purely in silico methods may not harness
ProteinMPNN and LigandMPNN to their best effectiveness. In addition, a
greatly expanded number of variants tested by wet lab will likely improve
our final results.
Our protein redesign pipeline produced
variants with significantly more negative ΔG values relative to the wildtype
structures. While we did not test the variants' half-life or reactivity at
dufferent temperatures, the current pipeline is a good starting point
for future stability-enhancement oriented design methods that will greatly
contribute to ArMOLDgeddon's commercial values by extending product shelf
life and increasing feasibility of product forms requiring long-term
enzymatic activity.
Acronyms
GlcNAc: N-acetylglucosamine, chitin is β-1,4-GlcNAc polymer
SASA: Solvent accessible surface area
REU: Rosetta energy unit
RMSD: Root mean square deviation
Supplementary Materials
Methods
batch_cif_to_pdb.py
import pathlib
import subprocess
def convert_cif_to_pdb(input_folder):
"""
Convert all CIF files in the specified folder to PDB format using OpenBabel.
The PDB files will be saved in the same folder.
Args:
input_folder (str): Path to the folder containing CIF files.
"""
folder_path = pathlib.Path(input_folder)
if not folder_path.exists():
print(f"Error: Folder '{folder_path}' does not exist.")
return
# Find all .cif files in the folder
cif_files = list(folder_path.glob("*.cif"))
if not cif_files:
print(f"No CIF files found in '{folder_path}'.")
return
print(f"Found {len(cif_files)} CIF file(s) to convert.")
successes = []
failures = []
for cif_file in cif_files:
pdb_file = cif_file.with_suffix(".pdb")
# OpenBabel command
cmd = [
"obabel",
"-icif", str(cif_file),
"-opdb", "-O", str(pdb_file),
"-e"
]
print(f"Converting '{cif_file.name}' to PDB...")
try:
# Run OpenBabel
subprocess.run(cmd, check=True, capture_output=True, text=True)
if pdb_file.exists() and pdb_file.stat().st_size > 0:
successes.append(cif_file.name)
print(f"Success: Saved as '{pdb_file.name}'")
else:
failures.append(cif_file.name)
print("Warning: No valid PDB file was created.")
except subprocess.CalledProcessError as e:
failures.append(cif_file.name)
print(f"Error: Failed to convert '{cif_file.name}': {e.stderr.strip()}")
except FileNotFoundError:
print("Error: 'obabel' command not found. Is OpenBabel installed?")
return
except Exception as e:
failures.append(cif_file.name)
print(f"Unexpected error converting '{cif_file.name}': {str(e)}")
# Summary
print("\nConversion Summary:")
print(f"Successful: {len(successes)}")
print(f"Failed: {len(failures)}")
if failures:
print("\nFailed files:")
for name in failures:
print(f"- {name}")
INPUT_FOLDER = r"C:\Users\Lenovo\pdb" #path to the input files (just write the path to folder, can handle all cif files in batch)
if __name__ == "__main__":
convert_cif_to_pdb(INPUT_FOLDER)
batch_fasta_to_json.py
import json
from pathlib import Path
def ligandmpnn_to_json_filtered(input_file_path, output_file=None):
"""
Parse LigandMPNN output file, OMIT the first sequence, and convert to JSON.
Uses the existing 'id' from the file for labeling.
"""
# Convert input path to Path object
input_path = Path(input_file_path)
if not input_path.exists():
print(f"Error: The file '{input_file_path}' does not exist.")
return
# create a file or just use the older one
if output_file is None:
output_file = input_path.with_suffix('.json')
try:
with open(input_path, 'r', encoding='utf-8') as f:
content = f.read()
# Parse the file and extract sequences only with ids specified (to avoid adding on the wt protien and waste valuable quota)
sequences_with_ids = [] # Store tuples of (sequence, id_from_file)
current_record = None
current_sequence = []
current_id = None
found_first_record = False
for line in content.split('\n'):
line = line.strip()
if line.startswith('>P1;'):
if current_record is not None and current_sequence and current_id is not None:
sequence_str = ''.join(current_sequence).replace(' ', '')
sequences_with_ids.append((sequence_str, current_id))
# repeat
current_record = line
current_sequence = []
# Check if this header contains an ID
if 'id=' in line:
# Extract the ID from the header
import re
id_match = re.search(r'id=(\d+)', line)
if id_match:
current_id = int(id_match.group(1))
else:
current_id = None
else:
# defined the first record (the wt) as one not having and id (for mpnn output this is usually the case)
current_id = None
found_first_record = True
elif line.startswith(', 0 氨基酸'): #apply to Chinese version of Snapgene fasta output
# Skip the metadata line
continue
elif line and current_record is not None:
# This is a sequence line - remove spaces and add to current sequence (the space is a formating in snapgene fasta file)
cleaned_line = line.replace(' ', '')
if cleaned_line and all(c in 'ACDEFGHIKLMNPQRSTVWY' for c in cleaned_line):
current_sequence.append(cleaned_line)
# Don't forget the last record (if it has an ID)
if current_record is not None and current_sequence and current_id is not None:
sequence_str = ''.join(current_sequence).replace(' ', '')
sequences_with_ids.append((sequence_str, current_id))
# Create JSON structure
json_data = []
base_name = input_path.stem # Get filename without extension
for sequence, original_id in sequences_with_ids:
entry = {
"name": f"{base_name}_{original_id}",
"modelSeeds": [],
"sequences": [
{
"proteinChain": {
"count": 1,
"sequence": sequence
}
}
],
"dialect": "alphafoldserver",
"version": 1
}
json_data.append(entry)
# Write to JSON file
with open(output_file, 'w', encoding='utf-8') as out_f:
json.dump(json_data, out_f, indent=4, ensure_ascii=False)
print(f"Successfully converted filtered LigandMPNN output to JSON")
print(f"Input: {input_path}")
print(f"Output: {output_file}")
print(f"Processed {len(sequences_with_ids)} sequences (first sequence omitted)")
# Show preview
if sequences_with_ids:
print(f"\nPreview of first processed entry:")
print(f" Name: {json_data[0]['name']}")
print(f" Original ID from file: {sequences_with_ids[0][1]}")
print(f" Sequence length: {len(sequences_with_ids[0][0])}")
print(f" First 50 chars: {sequences_with_ids[0][0][:50]}...")
except Exception as e:
print(f"Error during conversion: {str(e)}")
return 0
return len(sequences_with_ids)
input_file_path = r"C:\Users\Lenovo\file" #directly set the path to YOUR FILE, not folder
if __name__ == "__main__":
try:
num_sequences = ligandmpnn_to_json_filtered(input_file_path)
if num_sequences > 0:
print(f"\n Success! Your JSON file has been created with {num_sequences} sequences")
print(" The first sequence (without an 'id') has been omitted.")
else:
print("No sequences were processed")
except Exception as e:
print(f"Unexpected error: {str(e)}")
dock_schrodinger.py
import os
import glob
import argparse
import numpy as np
import subprocess
import sys
import tempfile
import time
import logging
from concurrent.futures import ProcessPoolExecutor
import multiprocessing
import pandas as pd # Import pandas for CSV integration
# Import Schrödinger specific modules (these require Schrödinger environment)
try:
from schrodinger import structure
from schrodinger.structutils import analyze
from schrodinger.structutils import rmsd
except ImportError:
# Handle cases where Schrödinger modules are not available (e.g., for testing parsing)
# Full script functionality will require a properly configured Schrödinger environment.
logging.warning("Schrödinger modules (structure, analyze, rmsd) not found. Script functionality will be limited.")
structure = None
analyze = None
rmsd = None
# Configure logging to file and console
logging.basicConfig(
level=logging.INFO, # Set to logging.DEBUG to see detailed argument types/values
format='%(asctime)s - %(processName)s - %(levelname)s - %(message)s',
handlers=[
logging.FileHandler('dock_schrodinger.log'),
logging.StreamHandler()
]
)
logger = logging.getLogger(__name__)
def parse_arguments():
"""Parse command-line arguments."""
parser = argparse.ArgumentParser(description="Dock native ligands into structures using Schrödinger API with parallel processing.")
parser.add_argument("structures_directory", help="Directory containing structure files (.pdb or .mae) with native ligands")
parser.add_argument("--max-workers", type=int, default=multiprocessing.cpu_count(),
help="Number of concurrent processes (default: number of CPU cores)")
parser.add_argument("--integrate-results", action="store_true",
help="If set, combine all individual *_glide.csv results into a single CSV file, excluding *_glide_skip.csv files.")
parser.add_argument("--reference-structure", type=str,
help="Optional: Path to a reference structure (.pdb or .mae) containing the ligand for RMSD calculation relative to docked poses.")
return parser.parse_args()
def load_structure(file_path):
"""Load a structure from a file, handling .pdb or .mae formats."""
if structure is None:
raise ImportError("Schrödinger 'structure' module not available. Cannot load structures.")
try:
reader = structure.StructureReader(file_path)
st = next(reader)
logger.info(f"Loaded structure from {file_path} with {len(st.atom)} atoms")
return st
except Exception as e:
logger.error(f"Failed to load structure from {file_path}: {e}")
raise
def extract_receptor_and_ligand(target_structure):
"""Extract the receptor and the first ligand from the structure."""
if analyze is None:
raise ImportError("Schrödinger 'analyze' module not available. Cannot extract receptor/ligand.")
try:
# This function finds a ligand based on common definitions.
ligands = analyze.find_ligands(target_structure)
if not ligands:
logger.warning("No ligand found in structure")
return None, None
# Take the first ligand found
ligand = ligands[0]
ligand_indexes = set(ligand.atom_indexes)
# Get all atom indexes in the structure
all_indexes = set(range(1, target_structure.atom_total + 1))
# Receptor is all atoms not part of the first ligand
receptor_indexes = list(all_indexes - ligand_indexes)
receptor_structure = target_structure.extract(receptor_indexes)
ligand_structure = ligand.st # The 'st' attribute is the structure.Structure object for the ligand
return receptor_structure, ligand_structure
except Exception as e:
logger.error(f"Failed to extract receptor and ligand: {e}")
raise
def prepare_receptor_with_prewizard(receptor_structure, prepwizard_script, options, output_file, base_name):
"""
Prepares the receptor using prepwizard_driver.py.
A temporary .mae file is created for input.
"""
temp_dir = tempfile.gettempdir()
# Create a unique temporary .mae file path for the receptor input
temp_input = os.path.join(temp_dir, f"{base_name}_temp_{os.getpid()}.mae")
try:
# Write the receptor structure directly to the temporary .mae file path.
# This resolves the issue where the file object 'f' was being passed
# to Schrodinger's structure.write, causing it to misinterpret the input.
receptor_structure.write(temp_input)
logger.info(f"Wrote temporary receptor file: {temp_input}")
# Add debugging for the types and values of arguments passed to subprocess
logger.debug(f"DEBUG: prepwizard_script type: {type(prepwizard_script)}, value: {prepwizard_script}")
logger.debug(f"DEBUG: temp_input type: {type(temp_input)}, value: {temp_input}")
logger.debug(f"DEBUG: output_file type: {type(output_file)}, value: {output_file}")
if not os.path.exists(temp_input):
raise FileNotFoundError(f"Temporary file not created: {temp_input}")
# Construct the command to run prepwizard_driver.py
# sys.executable ensures the script is run with the same Python interpreter
cmd = [sys.executable, prepwizard_script, temp_input, output_file] + options
logger.info(f"Running prepwizard_driver.py with command: {' '.join(cmd)}")
# Execute the subprocess
result = subprocess.run(cmd, check=True, capture_output=True, text=True)
logger.info(f"Protein preparation completed for {output_file}")
if result.stderr:
logger.warning(f"prepwizard_driver.py stderr: {result.stderr}")
# Verify if the output file was created
if not os.path.exists(output_file):
raise RuntimeError(f"Protein preparation failed: output file {output_file} not found")
except subprocess.CalledProcessError as e:
logger.error(f"Protein preparation failed with exit code {e.returncode}: {e.stderr}")
raise
finally:
# Attempt to clean up the temporary input file
max_attempts = 5
for attempt in range(max_attempts):
try:
if os.path.exists(temp_input):
os.remove(temp_input)
logger.info(f"Cleaned up temporary file: {temp_input}")
break
except PermissionError as e:
# Handle cases where the file might still be in use by another process
if attempt < max_attempts - 1:
logger.warning(f"Failed to delete {temp_input} (attempt {attempt + 1}/{max_attempts}): {e}")
time.sleep(1)
else:
logger.error(f"Failed to delete {temp_input} after {max_attempts} attempts: {e}")
def calculate_grid_center_and_size(ligand_structure):
"""Calculate grid center and box size based on the ligand."""
try:
coords = np.array([atom.xyz for atom in ligand_structure.atom])
centroid = coords.mean(axis=0)
distances = np.linalg.norm(coords - centroid, axis=1)
max_distance = distances.max()
# Add a buffer of 10 Å around the ligand
outer_box_size = 2 * max_distance + 10
return centroid, outer_box_size
except Exception as e:
logger.error(f"Failed to calculate grid parameters: {e}")
raise
def write_combined_inp(receptor_file, ligand_file, job_name, centroid, outer_box_size):
"""Write a combined .inp file for grid generation and docking."""
try:
inp_file = f"{job_name}.inp"
content = f"""
[ GRIDGEN ]
RECEP_FILE {receptor_file}
RECEP_VSCALE 1.0
GRID_CENTER {centroid[0]:.6f}, {centroid[1]:.6f}, {centroid[2]:.6f}
INNERBOX 3, 3, 3
OUTERBOX 39, 39, 39
[ DOCKING ]
LIGANDFILE {ligand_file}
PRECISION XP
EXPANDED_SAMPLING True
POSES_PER_LIG 10
LIG_VSCALE 0.8
WRITE_XP_DESC False
MAX_ITERATIONS 150
MAXKEEP 500000
MAXREF 4000
"""
with open(inp_file, 'w') as f:
f.write(content.strip())
logger.info(f"Generated combined input file: {inp_file}")
return inp_file
except Exception as e:
logger.error(f"Failed to write .inp file: {e}")
raise
def run_glide(inp_file, schrodinger_path, job_name):
"""Run Glide using the provided .inp file."""
try:
glide_cmd = os.path.join(schrodinger_path, 'glide')
# Added -JOBNAME flag with the job_name
args = [glide_cmd, inp_file, '-HOST', 'localhost:1', '-NJOBS', '1', '-JOBNAME', job_name, '-TMPLAUNCHDIR']
logger.info(f"Running Glide with command: {' '.join(args)}")
result = subprocess.run(args, text=True, check=True, capture_output=True)
logger.info(f"Glide completed for {inp_file}")
if result.stderr:
logger.warning(f"Glide stderr: {result.stderr}")
except subprocess.CalledProcessError as e:
logger.error(f"Glide failed for {inp_file} with exit code {e.returncode}: {e.stderr}")
raise
def integrate_glide_results(directory, rmsd_data):
"""
Finds all *_glide.csv files (excluding *_glide_skip.csv) in the specified directory
and combines them into a single CSV file, including RMSD data.
"""
logger.info(f"Integrating Glide results from directory: {directory}")
all_csv_files = glob.glob(os.path.join(directory, "*.csv"))
result_files = []
for csv_file in all_csv_files:
if csv_file.endswith("_glide.csv") and not csv_file.endswith("_glide_skip.csv"):
result_files.append(csv_file)
if not result_files:
logger.warning(f"No *_glide.csv files (excluding *_skip.csv) found for integration in {directory}.")
return
combined_df = pd.DataFrame()
for f in result_files:
try:
df = pd.read_csv(f)
combined_df = pd.concat([combined_df, df], ignore_index=True)
logger.info(f"Added {os.path.basename(f)} to combined results.")
except Exception as e:
logger.warning(f"Could not read {f} for integration: {e}")
if not combined_df.empty:
# Create a DataFrame from RMSD data
rmsd_df = pd.DataFrame(rmsd_data)
# Assuming 's_l_Title' in Glide CSV for joining, which should match the base_name
rmsd_df.rename(columns={'base_name': 's_l_Title'}, inplace=True)
# Merge combined_df with rmsd_df based on the ligand title
combined_df = pd.merge(combined_df, rmsd_df, on='s_l_Title', how='left')
output_path = os.path.join(directory, "combined_docking_results.csv")
combined_df.to_csv(output_path, index=False)
logger.info(f"Combined results saved to: {output_path}")
else:
logger.info("No data to combine. Combined results file not created.")
def process_structure(structure_file, prepwizard_script, options, schrodinger_path, reference_structure_path=None):
"""
Process a single structure: load, prepare, generate grid, and dock.
Returns a dictionary with base_name and calculated RMSD (or None).
"""
# Extract base name without extension
base_name = os.path.splitext(os.path.basename(structure_file))[0]
# Define a sub-directory for prepared structures relative to the input file
output_directory = os.path.join(os.path.dirname(structure_file), "prepared_structures")
os.makedirs(output_directory, exist_ok=True) # Ensure the directory exists
# Define absolute paths for output files
receptor_file = os.path.join(output_directory, f"{base_name}_receptor_prepared.maegz")
ligand_file = os.path.join(output_directory, f"{base_name}_ligand.maegz")
job_name = f"{base_name}_glide" # Glide job name, used for -JOBNAME flag
rmsd_value = None # Initialize RMSD value
try:
# Load input structure (original complex)
input_complex_st = load_structure(structure_file)
# Extract receptor and ligand from the input complex
receptor_st_input, ligand_st_input = extract_receptor_and_ligand(input_complex_st)
if receptor_st_input is None:
logger.warning(f"No ligand found in {structure_file}, skipping processing for this file.")
return {"base_name": base_name, "RMSD": None} # Return for integration even if skipped
# Prepare receptor using prepwizard
prepare_receptor_with_prewizard(receptor_st_input, prepwizard_script, options, receptor_file, base_name)
# Save the extracted ligand to an MAEGZ file
ligand_st_input.write(ligand_file)
logger.info(f"Saved ligand to {ligand_file}")
# Calculate grid center and size based on the ligand's coordinates
centroid, outer_box_size = calculate_grid_center_and_size(ligand_st_input)
# Write the combined .inp file for Glide
inp_file = write_combined_inp(receptor_file, ligand_file, job_name, centroid, outer_box_size)
# Run Glide, passing the job_name for the -JOBNAME flag
run_glide(inp_file, schrodinger_path, job_name)
# --- Optional RMSD Calculation ---
if reference_structure_path:
# Check if Schrödinger's RMSD module is available
if rmsd is None or analyze is None or structure is None:
logger.error("Schrödinger 'rmsd', 'analyze', or 'structure' module not available. Cannot calculate RMSD.")
elif not os.path.exists(reference_structure_path):
logger.error(f"Reference structure not found at '{reference_structure_path}'. Skipping RMSD calculation for {base_name}.")
else:
glide_output_maegz = f"{job_name}-out.maegz" # Standard Glide output name
if os.path.exists(glide_output_maegz):
try:
# Load the full docked complex (receptor + ligand) from Glide output
docked_complex_st = load_structure(glide_output_maegz)
# Load the full reference structure (receptor + ligand)
ref_overall_st = load_structure(reference_structure_path)
# Extract receptor and ligand from both structures
docked_receptor_st, docked_ligand_st = extract_receptor_and_ligand(docked_complex_st)
reference_receptor_st, reference_ligand_st = extract_receptor_and_ligand(ref_overall_st)
if docked_ligand_st and reference_ligand_st and docked_receptor_st and reference_receptor_st:
# 1. Superimpose the docked complex onto the reference complex based on protein atoms
# The transformation is calculated using the receptor structures
# We use atom_match_prop='element' for robust atom matching during superposition
# Ensure both receptors have atoms for superposition
if docked_receptor_st.atom_total > 0 and reference_receptor_st.atom_total > 0:
# Get transformation based on receptor alignment
# The returned xform applies to the 'moving_st' (docked_receptor_st here)
_, xform = rmsd.get_rmsd_and_superimpose_transformation(
docked_receptor_st, reference_receptor_st, atom_match_prop='element'
)
# Apply the transformation to the *entire* docked complex
# This creates a new structure where the protein is aligned to reference
docked_complex_st_aligned = xform.transform_structure(docked_complex_st)
# Extract the ligand from the newly aligned complex
_, docked_ligand_st_aligned = extract_receptor_and_ligand(docked_complex_st_aligned)
if docked_ligand_st_aligned:
# 2. Calculate RMSD of the docked ligand relative to the reference ligand
# after the protein-based alignment
rmsd_value = rmsd.get_rmsd(
docked_ligand_st_aligned, reference_ligand_st, atom_match_prop='element'
)
logger.info(f"RMSD of docked ligand relative to reference (protein-aligned) for {base_name}: {rmsd_value:.3f} Å")
else:
logger.warning(f"Could not re-extract ligand from aligned docked complex for {base_name}. Skipping RMSD calculation.")
else:
logger.warning(f"Receptor structures are empty for {base_name}. Skipping protein-aligned RMSD calculation.")
else:
logger.warning(f"Could not extract both receptor and ligand from either docked or reference structure for {base_name}. Skipping RMSD calculation.")
except Exception as e:
logger.error(f"Error encountered during protein-aligned RMSD calculation for {base_name}: {e}", exc_info=True)
else:
logger.warning(f"Glide output MAEGZ file not found at '{glide_output_maegz}' for RMSD calculation for {base_name}.")
# --- End Optional RMSD Calculation ---
logger.info(f"Completed processing for {structure_file}")
return {"base_name": base_name, "RMSD": rmsd_value} # Return the RMSD value for integration
except Exception as e:
logger.error(f"Error processing {structure_file}: {e}", exc_info=True)
# Return a dictionary with None for RMSD if an error occurs during processing
return {"base_name": base_name, "RMSD": None}
def main():
args = parse_arguments()
structures_directory = os.path.abspath(args.structures_directory)
max_workers = args.max_workers
reference_structure_path = args.reference_structure # Get the reference structure path
# Retrieve Schrödinger installation path from environment variable
schrodinger_path = os.environ.get('SCHRODINGER', '')
if not schrodinger_path:
logger.error("SCHRODINGER environment variable not set. Please set it to your Schrödinger installation directory.")
sys.exit(1)
# Construct the path to prepwizard_driver.py
# Assumes a standard Schrödinger installation structure
mmshare_dir = os.path.join(schrodinger_path, 'mmshare')
prepwizard_script = os.path.join(mmshare_dir, 'python', 'scripts', 'prepwizard_driver.py')
if not os.path.exists(prepwizard_script):
# Fallback to a common older path if the first one doesn't exist
mmshare_dir = os.path.join(schrodinger_path, 'mmshare-v6.9')
prepwizard_script = os.path.join(mmshare_dir, 'python', 'scripts', 'prepwizard_driver.py')
if not os.path.exists(prepwizard_script):
logger.error(f"prepwizard_driver.py not found at expected paths: {os.path.join(schrodinger_path, 'mmshare', 'python', 'scripts', 'prepwizard_driver.py')} or {os.path.join(schrodinger_path, 'mmshare-v6.9', 'python', 'scripts', 'prepwizard_driver.py')}. Please verify your SCHRODINGER environment variable and Schrödinger installation.")
sys.exit(1)
# Command-line options for prepwizard_driver.py
# These options configure how the protein is prepared (e.g., adding hydrogens, optimizing, etc.)
options = [
"-fillsidechains",
"-disulfides",
"-assign_all_residues",
"-rehtreat",
"-max_states", "1",
"-epik_pH", "7.4",
"-epik_pHt", "2.0",
"-antibody_cdr_scheme", "Kabat",
"-samplewater",
"-include_epik_states",
"-propka_pH", "7.4",
"-f", "S-OPLS",
"-rmsd", "0.3",
"-watdist", "5.0"
]
# Get all .pdb and .mae files from the specified directory
structure_files = glob.glob(os.path.join(structures_directory, "*.pdb")) + \
glob.glob(os.path.join(structures_directory, "*.mae"))
if not structure_files:
logger.warning(f"No .pdb or .mae files found in directory: {structures_directory}. Exiting.")
sys.exit(1)
# Process structures in parallel using a ProcessPoolExecutor
logger.info(f"Starting parallel processing for {len(structure_files)} structures with {max_workers} workers.")
# List to store RMSD results for integration
all_rmsd_results = []
with ProcessPoolExecutor(max_workers=max_workers) as executor:
futures = [
# Pass the reference_structure_path to process_structure
executor.submit(process_structure, structure_file, prepwizard_script, options, schrodinger_path, reference_structure_path)
for structure_file in structure_files
]
# Iterate over futures to catch and log any exceptions from worker processes
for future in futures:
try:
# Store the result from each process (which includes base_name and RMSD)
result_data = future.result()
if result_data: # Ensure result_data is not None (e.g., if a process totally failed)
all_rmsd_results.append(result_data)
except Exception as e:
logger.error(f"A task failed during parallel execution: {e}", exc_info=True) # exc_info=True logs traceback
# Integrate results if the --integrate-results flag is set
if args.integrate_results:
# Changed to os.getcwd() to look for CSVs in the terminal's working directory
integrate_glide_results(os.getcwd(), all_rmsd_results)
if __name__ == "__main__":
main()
superpose_pymol.py
# superpose_pymol.py
import os
import glob
import argparse
import numpy as np
import subprocess
import time
import sys
# Define a timeout
FILE_WAIT_TIMEOUT = 30
FILE_WAIT_INTERVAL = 1
def parse_arguments():
"""Parses command-line arguments for directory, reference structure."""
parser = argparse.ArgumentParser(description="Superpose PDB files onto a reference structure using PyMOL.")
parser.add_argument("input_directory", help="Directory containing PDB files to superpose")
parser.add_argument("ref_structure", help="Path to the reference structure file (e.g., .pdb or .mae)")
parser.add_argument("--output_directory", default=os.getcwd(),
help="Directory to save the superposed structure files. Defaults to current working directory.")
return parser.parse_args()
def load_structure_simple(file_path):
"""
Load a structure from a file for simple PyMOL use.
This is a basic placeholder; PyMOL handles the actual PDB parsing.
We just need to confirm file existence for input.
"""
if not os.path.exists(file_path):
raise FileNotFoundError(f"Structure file not found: {file_path}")
print(f"Confirmed existence of structure file: {file_path}")
# We don't actually parse it here, PyMOL will.
return file_path # Return path as confirmation
def _get_pymol_executable_path():
"""
Determines the best PyMOL executable path.
Prefers 'pymol.exe' (console) over 'PyMOLWin.exe' (GUI).
"""
base_pymol_path = r"C:\ProgramData\pymol" # Adjust this path if your PyMOL is installed elsewhere
pymol_console_exe = os.path.join(base_pymol_path, "pymol.exe")
pymol_win_exe = os.path.join(base_pymol_path, "PyMOLWin.exe")
if os.path.exists(pymol_console_exe):
print(f"Using console PyMOL executable: {pymol_console_exe}")
return pymol_console_exe
elif os.path.exists(pymol_win_exe):
print(f"Using GUI PyMOL executable (fallback): {pymol_win_exe}")
return pymol_win_exe
else:
raise FileNotFoundError(f"Neither '{pymol_console_exe}' nor '{pymol_win_exe}' found. Please verify PyMOL installation path in _get_pymol_executable_path().")
def superpose_structures_pymol(mobile_pdb_path, fixed_complex_pdb_path, output_aligned_pdb_path):
"""
Superpose a mobile protein structure onto the protein part of a fixed complex (protein+ligand)
using PyMOL's 'super' command, and then save the aligned mobile protein along with the ligand
from the fixed complex.
"""
fixed_complex_obj_name = "ref_complex"
mobile_protein_obj_name = "input_protein"
output_combined_obj_name = "aligned_complex_with_ligand"
# Convert all Windows backslashes to forward slashes for PyMOL commands
mobile_pdb_path_fwd = mobile_pdb_path.replace('\\', '/')
fixed_complex_pdb_path_fwd = fixed_complex_pdb_path.replace('\\', '/')
output_aligned_pdb_path_fwd = output_aligned_pdb_path.replace('\\', '/')
# PyMOL commands to be written into the .pml script
pymol_commands = [
f"load \"{fixed_complex_pdb_path_fwd}\", {fixed_complex_obj_name}",
f"load \"{mobile_pdb_path_fwd}\", {mobile_protein_obj_name}",
f"super {mobile_protein_obj_name}, {fixed_complex_obj_name} and polymer.protein",
f"create {output_combined_obj_name}, {mobile_protein_obj_name} or ({fixed_complex_obj_name} and not polymer.protein)",
f"save {str(output_aligned_pdb_path_fwd)}, {output_combined_obj_name}",
"quit" # Ensure PyMOL exits cleanly
]
command_string = "; ".join(pymol_commands)
# Get the appropriate PyMOL executable path
pymol_cmd_path = _get_pymol_executable_path()
# Create a temporary PyMOL script file to execute commands
temp_script_path = os.path.join(os.getcwd(), "temp_pymol_script.pml")
temp_script_path_fwd = temp_script_path.replace('\\', '/')
with open(temp_script_path, 'w') as f:
f.write(command_string)
# print("\n--- Diagnostic Information for PyMOL Execution ---")
# print(f"Python interpreter running script: {sys.executable}")
# print(f"PyMOL executable path used: {pymol_cmd_path}")
# print(f"Temporary PyMOL script path: {temp_script_path}")
# print(f"Mobile PDB (input) path: {os.path.abspath(mobile_pdb_path)}")
# print(f"Fixed Complex PDB (reference) path: {os.path.abspath(fixed_complex_pdb_path)}")
# print(f"Output Aligned PDB expected path: {os.path.abspath(output_aligned_pdb_path)}")
# print(f"Current Working Directory (cwd): {os.getcwd()}")
# print("--- Key Environment Variables for PyMOL Subprocess ---")
pymol_env = os.environ.copy()
pymol_base_dir = os.path.dirname(pymol_cmd_path)
pymol_env['PATH'] = pymol_base_dir + os.pathsep + pymol_env.get('PATH', '')
# print(f" PATH (for PyMOL subprocess) will start with: {pymol_base_dir}{os.pathsep}...")
# print("-------------------------------------------\n")
# print(f"Contents of temp_pymol_script.pml:\n{command_string}")
# print("-------------------------------------------\n")
full_command_args = [pymol_cmd_path, '-c', temp_script_path_fwd]
print(f"Attempting to execute PyMOL with command arguments: {full_command_args}")
try:
result = subprocess.run(full_command_args, text=True, check=True, cwd=os.getcwd(), capture_output=True, env=pymol_env)
print(f"PyMOL process finished with exit code {result.returncode}.")
if result.stdout:
print(f"PyMOL stdout:\n{result.stdout}")
if result.stderr:
print(f"PyMOL stderr:\n{result.stderr}")
print(f"Waiting for output file to appear: {output_aligned_pdb_path}")
file_found = False
start_time = time.time()
while time.time() - start_time < FILE_WAIT_TIMEOUT:
if os.path.exists(output_aligned_pdb_path) and os.path.getsize(output_aligned_pdb_path) > 0:
print(f"Output file found and is non-empty: {output_aligned_pdb_path}")
file_found = True
break
time.sleep(FILE_WAIT_INTERVAL)
if not file_found:
raise RuntimeError(f"PyMOL output file '{output_aligned_pdb_path}' was not found or remained empty after {FILE_WAIT_TIMEOUT} seconds.")
print(f"Successfully superposed {mobile_pdb_path} onto {fixed_complex_pdb_path} and saved to {output_aligned_pdb_path}")
return output_aligned_pdb_path
except FileNotFoundError:
raise RuntimeError(f"PyMOL executable not found at '{pymol_cmd_path}'. Please ensure the path is correct or 'pymol' is in your system PATH.")
except subprocess.CalledProcessError as e:
print(f"PyMOL process exited with error code {e.returncode}")
print(f"Command executed by subprocess: {' '.join(e.cmd)}")
if e.stdout:
print(f"PyMOL stdout (from error):\n{e.stdout}")
if e.stderr:
print(f"PyMOL stderr (from error):\n{e.stderr}")
raise RuntimeError(f"PyMOL superposition failed. See above output for details.")
except Exception as e:
raise RuntimeError(f"An unexpected error occurred during PyMOL execution: {e}")
finally:
if os.path.exists(temp_script_path):
os.remove(temp_script_path)
def main():
args = parse_arguments()
input_directory = os.path.abspath(args.input_directory)
ref_structure_path = os.path.abspath(args.ref_structure)
output_directory = os.path.abspath(args.output_directory)
os.makedirs(output_directory, exist_ok=True)
# Load reference structure (just check existence, PyMOL will do actual parsing)
fixed_pymol_ref = load_structure_simple(ref_structure_path)
pdb_files = glob.glob(os.path.join(input_directory, "*.pdb"))
if not pdb_files:
print(f"No .pdb files found in input directory: {input_directory}")
return
for pdb_file in pdb_files:
base_name = os.path.splitext(os.path.basename(pdb_file))[0]
output_aligned_pdb_path = os.path.join(output_directory, f"{base_name}_aligned_complex.pdb")
print(f"Processing {pdb_file} for superposition...")
try:
superpose_structures_pymol(
mobile_pdb_path=pdb_file,
fixed_complex_pdb_path=fixed_pymol_ref,
output_aligned_pdb_path=output_aligned_pdb_path
)
print(f"Finished superposition for {pdb_file}. Output saved to {output_aligned_pdb_path}")
except Exception as e:
print(f"Error superposing {pdb_file}: {e}")
continue
if __name__ == "__main__":
main()
Sumida, Kiera H., et al. "Improving Protein Expression, Stability, and
Function With ProteinMPNN." Journal of the American Chemical
Society, vol. 146, no. 3, Jan. 2024, pp. 2054–61,
doi:10.1021/jacs.3c10941.
Tyka, Michael D., et al. "Alternate States of Proteins Revealed by
Detailed Energy Landscape Mapping." Journal of Molecular Biology,
vol. 405, no. 2, Nov. 2010, pp. 607–18, doi:10.1016/j.jmb.2010.11.008.
Khatib, Firas, et al. "Algorithm Discovery by Protein Folding Game
Players." Proceedings of theNational Academy of
Sciences, vol. 108, no. 47, Nov. 2011, pp. 18949–53,
doi:10.1073/pnas.1115898108.
Maguire, Jack B., et al. "Perturbing the Energy Landscape for Improved
Packing During Computational Protein Design." Proteins Structure
Function and Bioinformatics, vol. 89, no. 4, Nov. 2020, pp. 436–49,
doi:10.1002/prot.26030.
Fleishman, Sarel J., Andrew Leaver-Fay, et al. "RosettaScripts: A
Scripting Language Interface to the Rosetta Macromolecular Modeling
Suite." PLoS ONE, vol. 6, no. 6, June 2011, p. e20161,
doi:10.1371/journal.pone.0020161.
Friesner, Richard A., et al. "Glide: A New Approach for Rapid, Accurate
Docking and Scoring. 1. Method and Assessment of Docking Accuracy."
Journal of Medicinal Chemistry, vol. 47, no. 7, Feb. 2004, pp.
1739–49, doi:10.1021/jm0306430.
Dauparas, J., et al. "Robust Deep Learning–based Protein Sequence Design
Using ProteinMPNN." Science, vol. 378, no. 6615, Sept. 2022, pp. 49–56,
doi:10.1126/science.add2187.
Dauparas, Justas, et al. "Atomic Context-conditioned Protein Sequence
Design Using LigandMPNN." Nature Methods, Mar. 2025,
doi:10.1038/s41592-025-02626-1.
Takashima, Tomoya, et al. "cDNA Cloning, Expression, and Antifungal
Activity of Chitinase From Ficus Microcarpa Latex: Difference in
Antifungal Action of Chitinase With and Without Chitin-binding Domain."
Planta, vol. 253, no. 6, May 2021,
doi:10.1007/s00425-021-03639-8.
Takashima, Tomoya, Ryo Sunagawa, et al. "Antifungal Activities of
LysM-domain Multimers and Their Fusion Chitinases." International
Journal of Biological Macromolecules, vol. 154, Nov. 2019, pp.
1295–302, doi:10.1016/j.ijbiomac.2019.11.005.
Huet, Joëlle, et al. "X-ray Structure of Papaya Chitinase Reveals the
Substrate Binding Mode of Glycosyl Hydrolase Family 19 Chitinases."
Biochemistry, vol. 47, no. 32, July 2008, pp. 8283–91,
doi:10.1021/bi800655u.
Gideon Davies, Nathalie Juge, and Vincent Eijsink, "Glycoside Hydrolase
Family 18" in CAZypedia, available at URL http://www.cazypedia.org /,
accessed 7 July 2025
Remmert, Michael, et al. "HHblits: Lightning-fast Iterative Protein
Sequence Searching by HMM-HMM Alignment." Nature Methods, vol. 9,
no. 2, Dec. 2011, pp. 173–75, doi:10.1038/nmeth.1818.
Abramson, Josh, et al. "Accurate Structure Prediction of Biomolecular
Interactions With AlphaFold 3." Nature, vol. 630, no. 8016, May
2024, pp. 493–500, doi:10.1038/s41586-024-07487-w.
Michino, Mayako, et al. "AI meets physics in computational
structure-based drug discovery for GPCRs." Npj Drug Discovery., vol. 2,
no. 1, July 2025, doi:10.1038/s44386-025-00019-0.
Miller, G. L. "Use of Dinitrosalicylic Acid Reagent for Determination of
Reducing Sugar." Analytical Chemistry, vol. 31, no. 3, Mar. 1959,
pp. 426–28, doi:10.1021/ac60147a030.
Trott, Oleg, and Arthur J. Olson. "AutoDock Vina: Improving the Speed
and Accuracy of Docking With a New Scoring Function, Efficient
Optimization, and Multithreading." Journal of Computational
Chemistry, vol. 31, no. 2, June 2009, pp. 455–61,
doi:10.1002/jcc.21334.
Nivedha, Anita K., et al. "Vina-Carb: Improving Glycosidic Angles During
Carbohydrate Docking." Journal of Chemical Theory and
Computation, vol. 12, no. 2, Jan. 2016, pp. 892–901,
doi:10.1021/acs.jctc.5b00834.
Jens Eklöf and Jan-Hendrik Hehemann, "Glycoside Hydrolase Family 16" in
CAZypedia, available at URL http://www.cazypedia.org/, accessed 12 July
2025
Goverde, Casper A., et al. "Computational Design of Soluble and
Functional Membrane Protein Analogues." Nature, vol. 631, no.
8020, June 2024, pp. 449–58, doi:10.1038/s41586-024-07601-y.
Onaga, Shoko, and Toki Taira. "A New Type of Plant Chitinase Containing
LysM Domains From a Fern (Pteris Ryukyuensis): Roles of LysM Domains in
Chitin Binding and Antifungal Activity." Glycobiology, vol. 18,
no. 5, Feb. 2008, pp. 414–23, doi:10.1093/glycob/cwn018.
Lee, Hyo-Jung, et al. "Cloning, Purification, and Characterization of an
Organic Solvent-tolerant Chitinase, MtCh509, From Microbulbifer
Thermotolerans DAU221." Biotechnology _for Biofu_els, vol. 11,
no. 1, Nov. 2018, doi:10.1186/s13068-018-1299-1.
King, Brianne R., et al. "Computational Stabilization of a Non‐heme Iron
Enzyme Enables Efficient Evolution of New Function." Angewandte Chemie
International Edition, Oct. 2024, doi:10.1002/anie.202414705.