Preview
The Cn2 and Css2 toxins, derived from the venom of the scorpions Centruroides noxius and Centruroides suffusus, are low molecular weight neurotoxic peptides (7.5 kDa) that bind to sodium channels, causing severe neurotoxic effects. (Universidad Autónoma del Estado de Morelos, Centro de Investigación en Biotecnología) (Universidad Autónoma del Estado de Morelos, Centro de Investigación en Biotecnología)., n.d.)
The Cn2 toxin structure has been experimentally resolved and is available in the Protein Data Bank (PDB ID: 1CN2), while Css2 was modeled by homology using its UniProt sequence. For the antibody scFv-LR, no experimentally solved structure is currently available, so a homology model was generated using SWISS-MODEL and AlphaFold. (Protein Data Bank, 2000) (Waterhouse et al., 2018; Jumper et al., 2021 & Jumper et al, 2018, )
In order to understand their interaction with a recombinant single-chain antibody fragment (scFv-LR), we performed structural modeling using UCSF Chimera 1.9, SWISS-MODEL, and AlphaFold. Three-dimensional models were obtained, prepared by the addition of hydrogens and assignment of partial charges using the AMBER ff14SB force field, followed by energy minimization. Key interactions were identified between scFv-LR and the toxins, including hydrogen bonds and electrostatic contacts with Asp7, Glu30, Lys31, Tyr36, Trp38, and Arg56 of Cn2. These results provide a reliable structural model that can serve as the foundation for subsequent molecular dynamics studies. (AMBER ff14SB., 2015)
Problem
Scorpion stings represent a significant public health issue in endemic regions of Mexico, where species of the Centruroides genus produce venoms containing neurotoxic peptides that act on sodium and calcium channels in neurons, leading to potentially lethal symptoms. (Possani Et Al, 2018)
The Cn2 (Centruroides noxius) and Css2 (Centruroides suffusus) toxins are among the most potent, consisting of 66–70 amino acids (7.5–8.0 kDa), stabilized by disulfide bridges. Current treatment relies on polyclonal antivenom sera, whose production is costly and associated with safety concerns. (Protein Data Bank, 2000)
The development of recombinant antibodies, such as single-chain variable fragments (scFvs), offers a safer and more specific alternative. Understanding how these antibodies interact with scorpion toxins is essential for optimizing their design. This work documents a complete structural modeling workflow to simulate and analyze the interaction between scFv-LR and the Cn2/Css2 toxins, using UCSF Chimera 1.9, SWISS-MODEL, and AlphaFold, aiming to provide a useful tool for IGEM and future therapeutic optimization. (AMBER ff14SB, 2015)
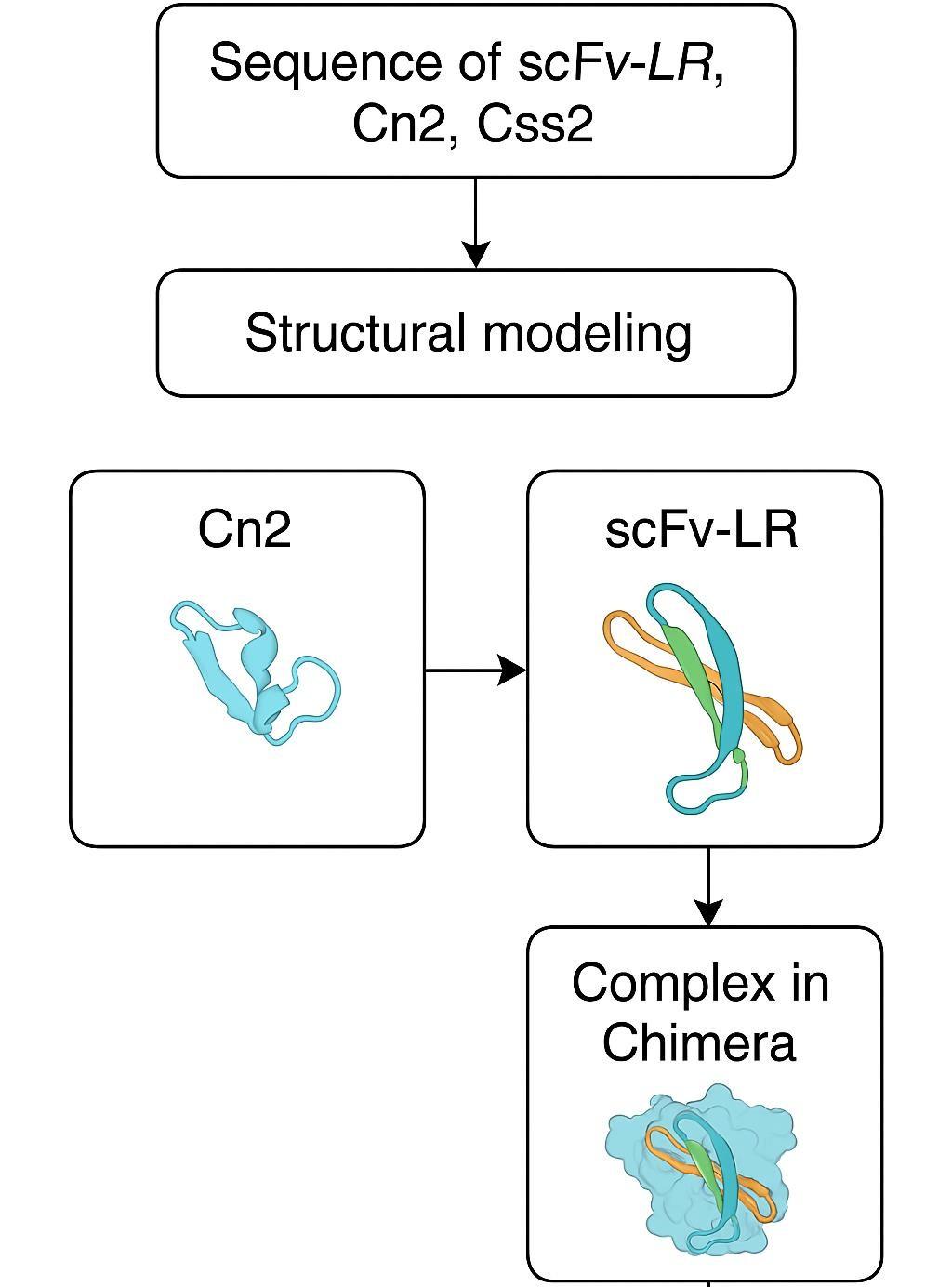
Figure 1: Schematic diagram of the overall workflow, from sequence acquisition to the final model.
Materials and Methods.
Structure acquisition:
- Cn2: downloaded from the Protein Data Bank (PDB ID: 1CN2).
- Css2: homology-modeled from its sequence (UniProt) using SWISS-MODEL.
- scFv-LR: sequence translated from DNA to amino acids and modeled using SWISS-MODEL/AlphaFold.
Figure 2: Three-dimensional structure of Cn2 rendered in Chimera.
Figure 3: Structure of scFv-LR with its VH and VL domains
Structure preparation in Chimera:
- Loading of .pdb files in Chimera 1.9.
- Addition of hydrogens using Tools - Structure Editing - AddH (simulation at physiological pH).
- Assignment of partial charges with Tools - Structure Editing - Add Charge (AMBER ff14SB force field).
- Positioning of scFv-LR near Cn2 and Css2 using movement tools.
- Energy minimization of the complex using Tools - Structure Editing - Minimize Structure (500 steps, gradient 0.02).
Cn2 and Css2 toxins share significant homology at both the sequence and structural level. Once the three-dimensional models were obtained, a structural alignment was performed in UCSF Chimera (MatchMaker tool). The results showed a proper overlap between the two toxins, with strong conservation in the β-sheet core and the characteristic α-helix of Centruroides toxins. This structural similarity suggests that both toxins present conserved epitopes, which is consistent with the ability of scFv-LR to recognize and potentially neutralize both in a similar manner.
Interaction analysis:
- Detection of contacts using Tools - Structure Analysis - Find Clashes/Contacts (maximum distance 3.5 Å).
- Identification of key residues: Asp7, Glu30, Lys31, Tyr36, Trp38, and Arg56 in Cn2.
Results
Accurate three-dimensional models of the toxins and scFv-LR were generated. Energy minimization produced a stable complex between the antibody and the Cn2 toxin, with multiple interactions: hydrogen bonds between the variable regions of the scFv and Asp7/Glu30, ionic interactions with Arg56, and hydrophobic contacts with Tyr36 and Trp38. For Css2, the model displayed a similar topology and binding mode, suggesting that scFv-LR could neutralize both toxins through a conserved mechanism. (Kozakov Et Al., 2017; Vajda Et Al., 2017; Jones Et Al., 2022, n.d.)
Molecular Docking Analysis with ClusPro
To validate and extend the observations made in UCSF Chimera, molecular docking was performed between the Cn2 toxin and the recombinant scFv-LR antibody using the ClusPro platform. scFv-LR was defined as the receptor and Cn2 as the ligand. The default “Balanced” mode was applied, which combines electrostatic and desolvation contributions.
ClusPro generated ten docking models (complex_1.pdb to complex_10.pdb), grouped into clusters according to structural similarity. The model complex_1.pdb exhibited the lowest weighted energy (−843.2 kcal/mol) and a cluster size of 75 members, suggesting high stability and reproducibility. (Kozakov Et Al., 2015, #)
The residues involved in the binding interface included Tyr33, Ser52, Trp103, and Arg98 of scFv-LR, interacting with Asp7, Glu30, and Trp38 of Cn2. These interactions were visualized in Chimera using pseudobonds and labels, and showed consistency with the residues previously identified manually.
The Balanced mode of ClusPro applies the following energy weighting function:
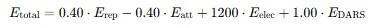
Important Considerations
This configuration prioritizes electrostatic contributions, which are essential for antigen–antibody interactions in peptide toxins such as Cn2, while still accounting for van der Waals forces and the statistical adjustment of the interface.
Table 1: ClusPro docking results: cluster size and weighted energy values.
| Cluster | Members | Representative | Weighted Score |
|---|---|---|---|
| 0 | 136 | Center | -643.0 |
| Lowest Energy | -784.3 | ||
| 1 | 135 | Center | -644.6 |
| Lowest Energy | -843.4 | ||
| 2 | 132 | Center | -789.6 |
| Lowest Energy | -865.5 | ||
| 3 | 112 | Center | -703.7 |
| Lowest Energy | -787.4 | ||
| 4 | 65 | Center | -640.4 |
| Lowest Energy | -686.3 | ||
| 5 | 43 | Center | -611.6 |
| Lowest Energy | -672.6 | ||
| 6 | 42 | Center | -620.9 |
| Lowest Energy | -698.9 | ||
| 7 | 41 | Center | -602.2 |
| Lowest Energy | -653.7 | ||
| 8 | 39 | Center | -631.9 |
| Lowest Energy | -690.4 | ||
| 9 | 39 | Center | -659.9 |
| Lowest Energy | -684.2 | ||
| 10 | 37 | Center | -646.7 |
| Lowest Energy | -647.8 |
*The column Cluster indicates the number assigned to each group of structurally similar models, while Members (size) refers to the number of models grouped within that cluster. The Center represents the energy of the model chosen as the representative of the cluster, and the Lowest-energy member corresponds to the lowest energy value among all models included in that cluster.
General form of the ClusPro docking energy equation
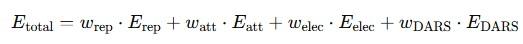
Term definitions:
- Etotal – Weighted total energy of the complex.
- Erep – Repulsive component of Van der Waals interactions (steric clashes).
- Eatt – Attractive component of Van der Waals interactions (dispersion forces).
- Eelec – Electrostatic interaction energy (between charges).
- EDARS – Statistical potential based on real protein–protein interfaces.
- Wrep – Weighting coefficients assigned to each term.
Balanced mode in ClusPro
In the Balanced mode, the weighting coefficients are defined as follows:
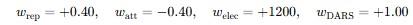
Therefore:
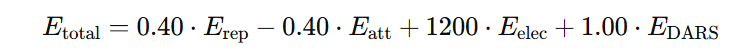
Example in Balanced mode.
In this work, the mode I used was:
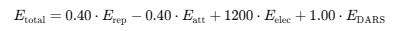
In the Balanced mode, the weighting coefficients are defined as follows:
- 0.40 for Erep – repulsion is considered but with moderate weight.
- −0.40 for Eatt – Van der Waals attraction lowers the total energy (favorable).
- +1200 for Eelec – electrostatics play an extremely important role in this model.
- +1.00 for EDARS – the statistical potential has a standard weight.
Why are these weights important?
The relative contribution of each term determines how the program prioritizes the predicted complexes:
- Biological relevance
In toxins such as Cn2, key interactions with antibodies are often electrostatic (e.g., Asp⁻ with Arg⁺). A high weight for Eelec (1200) ensures that ClusPro selects models where opposite charges are optimally aligned. (Kozakov Et Al., 2015, ) - Selection of the best model
If a Hydrophobic-favored mode had been used (zero weight for electrostatics and high weight for Van der Waals), the results would likely favor superficial non-specific interactions instead of blocking the functional epitope. (Kozakov Et Al., 2015) - Interpretation of results
Knowing that electrostatics dominate the scoring function allows us to interpret that short distances and favorable orientations of charged residues (Asp, Glu, Lys, Arg) are the most influential in the prediction. (Kozakov Et Al., 2015)
Application to scFv-LR – Cn2
- scFv-LR recognizes residues Asp7 and Glu30 (negatively charged) and Lys31 and Arg56 (positively charged) in Cn2.
- Due to the strong weighting of Eelec, ClusPro prioritized models in which the antibody charges complement those of the toxin.
- If electrostatics had a lower weight, the prediction could have positioned the antibody in non-functional regions of the toxin.
Table 2: Weighting coefficients and main applications of the different ClusPro docking modes.
| Docking Mode | Wrep | Watt | Welec | WDARS | Main application |
|---|---|---|---|---|---|
| Balanced (default) | +0.40 | −0.40 | +1200 | +1.00 | General protein–protein docking with balanced forces |
| Electrostatics-favored | 0.00 | 0.00 | +1500 | +1.00 | Systems dominated by charge interactions |
| Hydrophobic-favored | +1.00 | −1.00 | 0.00 | +1.00 | Interfaces stabilized mainly by hydrophobic contacts |
| Van der Waals + Elec | +0.40 | −0.40 | +600 | 0.00 | Classical physical interactions without statistical potential |
Note: Hydrogen bonds and polar contacts are mainly reflected in Eelec (due to orientation/charge) and partly in Eatt (distance/van der Waals adjustment). Hydrophobic interactions are mostly represented in Eatt; steric clashes are penalized in Erep. EDARS adds a “biological sense” to the interface based on structural statistics.
This table summarizes the weighting factors applied to each energy component in the ClusPro docking algorithm for its main docking modes. “Wrep,” “Watt,” “Welec,” and “WDARS” correspond to the weighting coefficients assigned to the repulsive Van der Waals, attractive Van der Waals, electrostatic, and statistical potential terms, respectively. The “Main application” column describes the typical use cases for each mode, highlighting how electrostatics, hydrophobicity, or balanced interactions influence the docking outcomes.
Table 3: . Energy components and physical meaning of the terms used in the ClusPro scoring function.
| Term | Meaning | Physical representation | Weight in the formula |
|---|---|---|---|
| Erep | Repulsive Van der Waals component (steric clashes) | Short-range Pauli repulsion due to electron-cloud overlap; prevents atomic interpenetration. | +0.40 |
| Eatt | Attractive Van der Waals component (dispersion forces) | London dispersion and general hydrophobic packing at optimal contact distances. | −0.40 |
| Eelec | Electrostatic interaction energy (charge–charge interactions) | Coulombic interactions: salt bridges (Arg/Lys–Asp/Glu), long-range charge attraction/repulsion; contributes to H-bonding orientation together with VdW. | +1200 |
| EDARS | Statistical potential based on real interfaces | Knowledge-based interface complementarity (residue–residue preferences learned from PPI datasets; “Decoys As the Reference State”). | +1.00 |
| Etotal | Weighted total energy of the complex | Overall scoring function combining the above contributions to rank docked poses. | Sum of all terms |
This table details the energetic terms that compose the ClusPro docking scoring function, their physical interpretation, and their relative weighting coefficients in the Balanced mode. “Erep” and “Eatt” correspond to the repulsive and attractive components of Van der Waals interactions, respectively; “Eelec” represents electrostatic interactions between charged residues; and “EDARS” is a knowledge-based statistical potential derived from real protein–protein interface data. “Etotal” corresponds to the overall weighted sum used to rank the docking poses according to their predicted stability.
What does the energy represent?
- Weighted energy (Etotal): A value in kilocalories per mole (kcal/mol) that summarizes:
- Electrostatic energy
- Desolvation energy (exposure to water)
- Repulsion energy
- The more negative the value, the stronger the predicted protein–protein binding.
Examples:
- −843.2 kcal/mol = excellent docking
- −600.0 kcal/mol = acceptable
- −300.0 kcal/mol = probably weak
(Kozakov Et Al., 2017; Vajda Et Al., 2017; Jones Et Al., 2022, n.d.)
Why did I choose these models?
I prioritized the model with:
- The most negative energy
- Largest cluster size (more models support it)
- Biological plausibility (interaction at the functional site)
Example:
- Cluster 1: 75 members, −843.2 kcal/mol → best model
- Cluster 3: 12 members, −750.0 kcal/mol → less reliable
Summary
The results indicate that scFv-LR recognizes critical epitopes in the toxins, most likely blocking their interaction sites with ion channels. The identification of key residues suggests opportunities to optimize affinity through targeted mutagenesis. This computational model serves as a starting point for molecular dynamics simulations with GROMACS and automated docking studies with HADDOCK or ClusPro, improving the prediction of binding affinities.
Conclusions
The workflow enabled detailed modeling and analysis of the interaction between scFv-LR and the toxins Cn2 and Css2. Careful preparation (addition of hydrogens and partial charges) and energy minimization ensured a reliable model, which will serve as the basis for further studies within the IGEM UAM project, including experimental validation and optimization of recombinant antivenoms.
References
- Ahmad, Z. A., Yeap, S. K., Ali, A. M., Ho, W. Y., Alitheen, N. B., & Hamid, M. (2012). scFv antibody: Principles and clinical application. Clinical and Developmental Immunology, 2012, 980250. https://doi.org/10.1155/2012/980250
- Chippaux, J. P. (2017). Emerging options for the management of scorpion stings. Drug Design, Development and Therapy, 11, 117–128. https://doi.org/10.2147/DDDT.S118265
- Desta, I. T., Porter, K. A., Xia, B., Kozakov, D., & Vajda, S. (2020). Performance and its limits in rigid-body protein–protein docking. Structure, 28(9), 1071–1081. https://doi.org/10.1016/j.str.2020.06.006
- Guzmán Flores, F. (s.f.). Expresión del anticuerpo scFv 6009F con el vector pHIL-S1 en Pichia pastoris. Universidad Autónoma del Estado de Morelos, Centro de Investigación en Biotecnología.
- Jones, G., Jindal, A., Ghani, U., Kotelnikov, S., Egbert, M., Hashemi, N., Vajda, S., Padhorny, D., & Kozakov, D. (2022). Elucidating protein function through computational docking and hotspot analysis by ClusPro and FTMap. Acta Crystallographica Section D: Structural Biology, 78(6), 690–697. https://doi.org/10.1107/S2059798322004592
- Jumper, J., Evans, R., Pritzel, A., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
- Kozakov, D., Beglov, D., Bohnuud, T., Mottarella, S., Xia, B., Hall, D. R., & Vajda, S. (2013). How good is automated protein docking? Proteins: Structure, Function, and Bioinformatics, 81(12), 2159–2166. https://doi.org/10.1002/prot.24403
- Kozakov, D., Hall, D. R., Xia, B., Porter, K. A., Padhorny, D., Yueh, C., Beglov, D., & Vajda, S. (2017). The ClusPro web server for protein–protein docking. Nature Protocols, 12(2), 255–278. https://doi.org/10.1038/nprot.2016.169
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. https://doi.org/10.1002/jcc.20084
- Possani, L. D., et al. (2018). Scorpion toxins specific for Na+-channels. European Journal of Pharmacology, 834, 5–19. https://doi.org/10.1016/j.ejphar.2018.07.026
- Protein Data Bank. (2000). Crystal structure of scorpion toxin Cn2 (PDB ID: 1CN2). RCSB PDB. https://www.rcsb.org/structure/1CN2
- SWISS-MODEL: Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T. A. P., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303. https://doi.org/10.1093/nar/gky427
- Vajda, S., Yueh, C., Beglov, D., Bohnuud, T., Mottarella, S. E., Xia, B., Hall, D. R., & Kozakov, D. (2017). New additions to the ClusPro server motivated by CAPRI. Proteins: Structure, Function, and Bioinformatics, 85(3), 435–444. https://doi.org/10.1002/prot.25219